+
+
+
+Content not found. Please use links in the navbar.
+
+ +> Fix End of Files.........................................................Passed
+> Trim Trailing Whitespace.................................................Failed
+> - hook id: trailing-whitespace
+> - exit code: 1
+> - files were modified by this hook
+>
+> Fixing path/to/changed/files/file.txt
+>
+> codespell................................................................Passed
+> style-files..........................................(no files to check)Skipped
+> readme-rmd-rendered..................................(no files to check)Skipped
+> use-tidy-description.................................(no files to check)Skipped
+ +In the example above, one of the hooks modified a file in the proposed commit, +so the pre-commit check failed. You can run `git diff` to see the changes that +pre-commit made and `git status` to see which files were modified. To proceed +with the commit, re-add the modified file(s) and re-run the commit command: + +```sh +git add path/to/changed/files/file.txt +git commit -m 'feat: create function for awesome feature' +``` + +This time, all the hooks either passed or were skipped +(e.g. hooks that only run on R code will not run if no R files were +committed). +When the pre-commit check is successful, the usual commit success message +will appear after the pre-commit messages showing that the commit was created. + +> Check for added large files..............................................Passed
+> Fix End of Files.........................................................Passed
+> Trim Trailing Whitespace.................................................Passed
+> codespell................................................................Passed
+> style-files..........................................(no files to check)Skipped
+> readme-rmd-rendered..................................(no files to check)Skipped
+> use-tidy-description.................................(no files to check)Skipped
+> Conventional Commit......................................................Passed
+> [iss-10 9ff256e] feat: create function for awesome feature
+> 1 file changed, 22 insertions(+), 3 deletions(-)
+ +Finally, push your changes to GitHub: + +```sh +git push +``` + +If this is the first time you are pushing this branch, you may have to +explicitly set the upstream branch: + +```sh +git push --set-upstream origin iss-10 +``` + +> Enumerating objects: 7, done.
+> Counting objects: 100% (7/7), done.
+> Delta compression using up to 10 threads
+> Compressing objects: 100% (4/4), done.
+> Writing objects: 100% (4/4), 648 bytes | 648.00 KiB/s, done.
+> Total 4 (delta 3), reused 0 (delta 0), pack-reused 0
+> remote: Resolving deltas: 100% (3/3), completed with 3 local objects.
+> remote:
+> remote: Create a pull request for 'iss-10' on GitHub by visiting:
+> remote: https://github.com/CCBR/SCWorkflow/pull/new/iss-10
+> remote:
+> To https://github.com/CCBR/SCWorkflow
+>
+> [new branch] iss-10 -> iss-10
+> branch 'iss-10' set up to track 'origin/iss-10'.
+ +We recommend pushing your commits often so they will be backed up on GitHub. +You can view the files in your branch on GitHub at +`https://github.com/CCBR/SCWorkflow/tree/
+
The Single Cell Workflow streamlines the analysis of multimodal Single Cell RNA-Seq data produced from 10x Genomics. It can be run in a docker container, and for biologists, in user-friendly web-based interactive notebooks (NIDAP, Palantir Foundry). Much of it is based on the Seurat workflow in Bioconductor, and supports CITE-Seq data. It incorporates a cell identification step (ModScore) that utilizes module scores obtained from Seurat and also includes Harmony for batch correction. -Some of the steps in the workflow: +## Key Functions + +### Sequential Workflow +1. **processRawData()** - Process H5 files into Seurat objects +2. **filterQC()** - Quality control and filtering +3. **combineNormalize()** - Merge samples, normalize, dimension reduction +4. **Harmony integration** (optional) - Batch correction +5. **annotateCellTypes()** - Automatic cell type annotation via SingleR + +### Analysis & Visualization +- **compareCellPopulations()** - Compare cell population distributions across groups +- **degGeneExpressionMarkers()** - Differential expression analysis +- **reclusterSeuratObject()** / **reclusterFilteredSeuratObject()** - Subset and re-cluster +- **colorByGene()**, **heatmapSC()**, **violinPlot()** - Visualization functions +- **plotMetadata()**, **dotPlotMet()** - Metadata visualization + +
+ +For further documentation see our detailed [Docs Website](https://nidap-community.github.io/SCWorkflow/) -
 +
++
Future Developments include addition of support for multiomics (TCR-Seq, ATAC-Seq) single cell data and integration with spatial transcriptomics data. diff --git a/_pkgdown.yml b/_pkgdown.yml new file mode 100644 index 0000000..caab5bc --- /dev/null +++ b/_pkgdown.yml @@ -0,0 +1,77 @@ +url: https://nidap-community.github.io/SCWorkflow/ +template: + bootstrap: 5 + bslib: + primary: "#296b7f" + secondary: "#7cc349" + base_font: {google: "Roboto"} + heading_font: {google: "Roboto"} + code_font: {google: "Roboto Mono"} + border-radius: 0 + btn-border-radius: 3px + grid-gutter-width: 3rem + pkgdown-nav-height: 78px + params: + toc: + smooth_scroll: true + toc_float: true + toc_collapsed: true # Start with TOC collapsed + +navbar: + structure: + left: [getting-started, articles, Developers, reference] + right: [search, github] + components: + getting-started: + text: Getting Started + href: articles/SCWorkflow-Usage.html + Developers: + text: For Developers + href: articles/CONTRIBUTING.html + + +footer: + structure: + left: [developed_by] + +development: + mode: auto +authors: + Kelly Sovacool: + href: "https://github.com/kelly-sovacool" + Philip Homan: + href: "https://github.com/phoman14" + Vishal Koparde: + href: "https://github.com/kopardev" + CCR Collaborative Bioinformatics Resource: + href: "https://github.com/CCBR" + footer: + roles: [cph, fnd] + text: "Created by the" + +articles: +- title: Vignettes + navbar: ~ + contents: + - SCWorkflow-QC + - SCWorkflow-Annotations + - SCWorkflow-DEG + - SCWorkflow-Visualizations + - SCWorkflow-SubsetReclust + +- title: Developer + desc: Developer documentation + contents: + - CONTRIBUTING + - SCWorkflow-Usage + - README + +home: + sidebar: + structure: [links,citation, authors, license, dev] + links: + - text: Browse code on GitHub + href: https://github.com/NIDAP-Community/SCWorkflow + strip_header: true + + diff --git a/docs/404.html b/docs/404.html new file mode 100644 index 0000000..f371895 --- /dev/null +++ b/docs/404.html @@ -0,0 +1,89 @@ + + + + + + + +
+
+
+
+
+
+
+
+
diff --git a/docs/CHANGELOG.html b/docs/CHANGELOG.html
new file mode 100644
index 0000000..d7caeea
--- /dev/null
+++ b/docs/CHANGELOG.html
@@ -0,0 +1,821 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/LICENSE-text.html b/docs/LICENSE-text.html
new file mode 100644
index 0000000..f0f2a04
--- /dev/null
+++ b/docs/LICENSE-text.html
@@ -0,0 +1,67 @@
+
+
+
+
+
+
+
+
+
+CHANGELOG
+ +
+
+
+
+
+
+v1.0.3 (in development)
+
+
+Feature
+- feat: Add compareCellPopulations() function for comparing cell population distributions across experimental groups
+
- Visualizes cell population frequencies or absolute counts across multiple groups +
- Generates alluvial flow bar plots and faceted box plots +
- Supports custom group ordering and color palettes +
- Added ggalluvial dependency for flow visualizations +
- Generated from JSON template using json2r.prompt.md instructions +
+
+
+v1.0.2 (2024-02-01)
+
+
+
+Fix
+- fix: update package name in action file (
00c816c)
+
+
+v1.0.1 (2024-02-01)
+
+
+Build
+- build: update conda recipe and action file (
32f800c)
+
+
+
+
+
+Documentation
+docs(version): Automatic development release (
0548007)
+docs: Adding changelog (
34b1e0b)
+docs(version): Automatic development release (
184b4f9)
+docs(version): Automatic development release (
b8b41d3)
+docs(version): Automatic development release (
f420a3b)
+docs(version): Automatic development release (
8b5cc98)
+
+
+Unknown
+- Merge pull request #51 from NIDAP-Community/dev +
Update test files for Harmony and Annotation, add GitHub page image (5c19124)
- Merge pull request #50 from NIDAP-Community/CDupdate +
fix: add skip on CI for harmony (3268a82)
- Merge pull request #49 from NIDAP-Community/CDupdate +
fix: Suppress warning for celldex, move CI handle for test Harmony, a… (30de884)
- Merge pull request #47 from NIDAP-Community/dev +
Update CD (76e59b4)
- Merge pull request #46 from NIDAP-Community/CDupdate +
docs: Adding changelog (7c5bcf2)
- Merge pull request #45 from NIDAP-Community/dev +
Update repo structure for CD implementation (ca27382)
- Merge pull request #44 from NIDAP-Community/CDupdate +
Update repo structure for Continuous Deployment implementation (fab9424)
Merge remote-tracking branch 'origin/main' into dev (
c850a61)
+Merge pull request #43 from NIDAP-Community/revert-42-testCD2
+
Revert "Test cd2" (dbd1511)
Revert "Test cd2" (
82c3208)
+Merge pull request #42 from ruiheesi/testCD2
+
Test cd2 (696718c)
- Merge pull request #17 from ruiheesi/release_dev +
Release dev (93dac20)
- Merge pull request #16 from ruiheesi/dev +
Dev (f094e92)
- Merge pull request #15 from ruiheesi/testCD2 +
Test cd2 (89850c1)
Merge remote-tracking branch 'origin/dev' into testCD2 (
429581c)
+feat :test (
10b3e05)
+Merge pull request #14 from ruiheesi/dev
+
Dev (c932c71)
- Merge pull request #13 from ruiheesi/testCD2 +
Test cd2 (da01ac7)
- Merge pull request #12 from ruiheesi/release_dev +
Release dev (447457a)
- Merge pull request #11 from ruiheesi/dev +
Dev (e4a2987)
- Merge pull request #10 from ruiheesi/testCD2 +
fix: update readme (a3f0d9c)
- Merge pull request #9 from ruiheesi/release_dev +
Release dev (4026e1a)
- Merge pull request #8 from ruiheesi/dev +
Dev (4364548)
- Merge pull request #7 from ruiheesi/testCD +
test: update meta.ymal (fe470f9)
- Merge pull request #6 from ruiheesi/dev +
Dev (4e75a9f)
- Merge pull request #5 from ruiheesi/testCD +
test: update meta.ymal (caed439)
- Merge pull request #4 from ruiheesi/dev +
Dev (9f27de1)
- Merge pull request #3 from ruiheesi/testCD +
test: mute line42 in test-Process_Raw_Data (abef880)
- Merge pull request #1 from ruiheesi/testCD +
feat: enable CD (0194dd9)
- Merge pull request #38 from NIDAP-Community/main +
Updating dev to avoid potential lost of progress (8936388)
- Merge pull request #37 from NIDAP-Community/8_4_tutorial +
8 4 tutorial (c640c8f)
Merge branch '8_4_tutorial' of https://github.com/NIDAP-Community/SCWorkflow into 8_4_tutorial (
be8b11f)
+Just for tutorial (
b667bb0)
+test (
d8bc63c)
+Fixing visualization for "after" plot (
d7bb90f)
+Merge pull request #36 from NIDAP-Community/dev
+
Dev (0b66085)
- Merge pull request #35 from NIDAP-Community/heatmap_fix +
Fix heatmap (49f0486)
Fix heatmap (
db3ee4e)
+Merge pull request #34 from NIDAP-Community/release_6_15_test
+
Update DESCRIPTION file with author info and short package description (31a4d13)
- Merge pull request #33 from NIDAP-Community/update_DES +
Update DESCRIPTION file (df2abd7)
Update DESCRIPTION file (
5e43253)
+Adding auto-generated files (
14ff346)
+Merge pull request #31 from NIDAP-Community/release_6_13
+
Update 6 13 from Alexei (e2297c6)
- Merge pull request #30 from NIDAP-Community/release_test +
Run all unit tests (407ca8f)
- Merge pull request #28 from NIDAP-Community/main +
Update (2891614)
- Merge pull request #27 from NIDAP-Community/phil_6_6_no_NG +
Modify package function load (370522e)
Including "NULL" and "seurat_cluster" tests (
8cb6a23)
+Introducing "cluster" variable functionality (
e26f2aa)
+Modify package function load (
fe7ad65)
+Adding auto-generated files (
e65ad6b)
+Merge pull request #26 from NIDAP-Community/release_test
+
Passed all tests (4a40802)
- Merge pull request #25 from NIDAP-Community/phil_6_6_no_NG +
Run all unit test (4979266)
Update Plot_Metadata (
573b57a)
+Update ModuleScore (
4367b28)
+Update NAMESPACE (
77240ed)
+Merge changes (
b9226e0)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
Conflicts: NAMESPACE tests/testthat/fixtures/downsample_SO.R (d4b92fe)
update documentation (
19b7179)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
ca74b58)
+delete old files (
b5b2d1d)
+Removed test-Pseudobulk_DEG.R and test-Sample_Names.R (
877fa28)
+Removed test-Meta_Data.R (
975face)
+Merge branch 'main' into phil_6_6_no_NG (
f638db9)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
77c1f03)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
need to update violinPlot subset function (41f805e)
update violinPlot subset function (
0609899)
+Remove old tests (
6ca9164)
+udate PBMC sing Filtered rds (
516a9df)
+Fix Test Error (
7641655)
+fix Test error (
e97494a)
+Merge branch 'main' into phil_6_6_no_NG (
d7fd7c8)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
1e52f51)
+Setting add.gene.or.protein to TRUE - will error out on FALSE (
2e0a153)
+Merge branch 'main' into phil_6_6_no_NG (
d61ce1d)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
cd90fbb)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
b44581b)
+bug fix on heatmap sort by annotation (
8d09781)
+Merge branch 'main' into phil_6_6_no_NG (
986bcdf)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
8fab93f)
+changed select_crobject to selectCRObject (
b7446a6)
+Update Seurate importing method in process raw (
846ac5d)
+Trigger check and update latest main (
2b575eb)
+Merge branch 'main' into phil_6_6_no_NG (
aab0562)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
b786ed1)
+Removed gene from Seurat (
d5d17ac)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
e7f08eb)
+Bug fix for dual labeling (
9eede5b)
+Fixed bug in Dual Labeling and added one more plot output (contingency table) (
0b17248)
+Test direct import seurat in process raw (
3bd763b)
+Trigger action, updated process raw to import Seurat (
2e46082)
+Trigger action, updated cc.gene (
84acd38)
+Trigger action, updated NAMESPACE (
a968d2d)
+Trigger action (
b38dd32)
+Trigger action (
05e9be4)
+Update NAMESPACE (
d7015e6)
+Merge branch 'main' into phil_6_6_no_NG (
d9ba725)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
5559288)
+Trigger unit tests (
99ae357)
+Add Filtered rds (
666c3d8)
+update Variable descriptions (
43e05f5)
+update Variable descriptions (
3fd169b)
+reverting from clusters to barcodes for merging (
f561e2f)
+Trigger unit test (
a668ef9)
+Trigger unit test (
1c48c37)
+Merge branch 'main' into phil_6_6_no_NG (
ada4aab)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
aa254f1)
+Update NAMESPACE (
dc6152b)
+rn h5 test (
d398586)
+Fixing "All" cluster label (
18bbdc8)
+Removing "latent variable" from a test script (
1839cdd)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
Conflicts: NAMESPACE R/Combine_and_Renormalize.R R/Filter_and_QC.R R/PCA_and_Normalization.R R/Post_filter_QC_Plots.R tests/testthat/fixtures/NSCLC_Single/NSCLC_Single_Filtered_PCA_Norm_SO_downsample.rds tests/testthat/fixtures/NSCLC_Single/NSCLC_Single_Filtered_SO_downsample.rds tests/testthat/fixtures/NSCLC_Single/NSCLCsingle_Filtered_PCA_Norm_SO_downsample.rds tests/testthat/fixtures/NSCLC_Single/NSCLCsingle_Filtered_SO_downsample.rds tests/testthat/fixtures/PBMC_Single/PBMC_Single_Filtered_PCA_Norm_SO_downsample.rds tests/testthat/fixtures/PBMC_Single/PBMC_Single_Filtered_SO_downsample.rds tests/testthat/fixtures/downsample_SO.R tests/testthat/test_Combine_and_Renormalize.R tests/testthat/test_Filter_and_QC.R tests/testthat/test_PCA_and_Normalization.R tests/testthat/test_Post_Filter_QC.R (92eae54)
FiltQC Variable Descriptions (
4aa0674)
+New RAW filtQC CombNorm (
7aa1a24)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
Conflicts: NAMESPACE R/Post_filter_QC_Plots.R tests/testthat/test_Post_Filter_QC.R (4fd27d2)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
Conflicts: man/Combine_and_Renormalize.Rd man/Post_filter_QC.Rd (159af06)
skip on ci on reticulate pacakge (
549f205)
+skip on ci on reticulate pacakge (
75412d7)
+Edited snapshot tests (
a316ad3)
+Edited snapshot tests (
cf00505)
+update pseudobulk helper (
fd0a506)
+update pseudobulk helper (
7f8060e)
+add pseudobulk helper and test scripts (
122f072)
+add pseudobulk helper and test scripts (
27a49ec)
+update functions and tests (
c7ddbf8)
+update functions and tests (
c201dd7)
+Removing "latent var" and replacing second.clust (
67c3aea)
+Removing "latent var" and replacing second.clust (
9c16231)
+Quick test for CI (
e71339c)
+Quick test for CI (
73c62ca)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
b600bf0)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
8bb7502)
+Added color selection to 3D-tsne Plotter (
2882b76)
+Added color selection to 3D-tsne Plotter (
f7decbc)
+Added a "cached" CellDex option (
2cabb8d)
+Added a "cached" CellDex option (
c913672)
+fixed NAMESPACE conflicts (
f2aa02f)
+fixed NAMESPACE conflicts (
797ab6d)
+Changed pheatmap to ComplexHeatmap::pheatmap (
5c9316b)
+Changed pheatmap to ComplexHeatmap::pheatmap (
414723d)
+Code Review (
03f4392)
+Code Review (
b2724c8)
+Removed LICENCE file in the description (
e2affa9)
+Removed LICENCE file in the description (
8c3154e)
+Minor fixes for R CMD CHECK (
134523f)
+Minor fixes for R CMD CHECK (
c1eb410)
+Fix syntax error
+
At line 200, fix syntax error from "=" to "==" (2916381)
- Fix syntax error +
At line 200, fix syntax error from "=" to "==" (d909584)
Helper for Recluster, Sprint 7 compliant. (
d5181f9)
+Helper for Recluster, Sprint 7 compliant. (
8f28252)
+New test for Recluster with correct name. (
bf252e2)
+New test for Recluster with correct name. (
0d88348)
+Old file w bad name gone. New file good. (
e903ca0)
+Old file w bad name gone. New file good. (
0e0d81e)
+Re-arranged for new Sprint 7 formatting and functions. (
2c07bcb)
+Re-arranged for new Sprint 7 formatting and functions. (
e670fe9)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
need to push updated code to main (20f0ab7)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
need to push updated code to main (189075c)
push updated scripts to main (
ed4b682)
+push updated scripts to main (
f70b090)
+Restructured according to new requirements (
94b0170)
+Restructured according to new requirements (
797d5be)
+Restructured according to new requirements (
495be93)
+Restructured according to new requirements (
7e53872)
+Restructured according to new requirements (
6043df1)
+Restructured according to new requirements (
5efd01d)
+Restructured according to new requirements (
8465a9e)
+Restructured according to new requirements (
75be9d0)
+Restructured according to new requirements (
8c4739b)
+Restructured according to new requirements (
8a39e82)
+Restructured according to new requirements (
2ccad56)
+Restructured according to new requirements (
4770cf7)
+Restructured according to new requirements (
8762e39)
+Restructured according to new requirements (
c21ca07)
+Restructured according to new requirements (
b9dfed4)
+Restructured according to new requirements (
9cc27d3)
+Restructured according to new requirements (
656b12e)
+Restructured according to new requirements (
2989dbe)
+Restructured according to new requirements (
bf8dcb1)
+Restructured according to new requirements (
d11a692)
+Restructured according to new requirements (
95693f6)
+Restructured according to new requirements (
da04b19)
+Restructured according to new requirements (
6dca0bf)
+Restructured according to new requirements (
de6b4da)
+Restructured according to new requirements (
8f4ea59)
+Restructured according to new requirements (
1741342)
+Restructured according to new requirements (
f522e45)
+Restructured according to new requirements (
dc8cfe2)
+Restructured according to new requirements (
b2235b0)
+Restructured according to new requirements (
6f4bd6d)
+Delete test-DegGeneExpressionMarkers.R
+
Replaced with newer, renamed version (237eacf)
- Delete test-DegGeneExpressionMarkers.R +
Replaced with newer, renamed version (ee510b7)
Accepted changes to Namespace (
b0ef9e7)
+Accepted changes to Namespace (
70d880f)
+Removed ticks on dotplot code (
d6599be)
+Removed ticks on dotplot code (
3b4a6e0)
+Added select to dplyr import (
1311aae)
+Added select to dplyr import (
c860639)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
8759676)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
28e310b)
+Added snapshot file tests to Name clusters (
1a0c530)
+Added snapshot file tests to Name clusters (
de98847)
+Added more snapshot tests to heatmap (
3f138a4)
+Added more snapshot tests to heatmap (
1fe2d7b)
+Additional snapshot file tests for dual labeling (
8d92fcf)
+Additional snapshot file tests for dual labeling (
580ab8d)
+Additional tests to dotplot by metadata (
fbc3ada)
+Additional tests to dotplot by metadata (
b7f6c45)
+Adding expect snapshot file tests to 3D-tSNE plotter (
8f33e2b)
+Adding expect snapshot file tests to 3D-tSNE plotter (
661102c)
+Formatting changes to helper-dual labeling (
8f5da99)
+Formatting changes to helper-dual labeling (
266dec8)
+Formatting changes to helper - Name clusters (
5a6079f)
+Formatting changes to helper - Name clusters (
cfc8b69)
+Formatting changes to helper-Heatmap (
abd3f09)
+Formatting changes to helper-Heatmap (
1ca0e49)
+Formatting changes to helper-dotplot by Metadata (
321e619)
+Formatting changes to helper-dotplot by Metadata (
69653cb)
+Formatting changes to helper 3D-tSNE (
8be697c)
+Formatting changes to helper 3D-tSNE (
c1835c2)
+Formatting changes to Dotplot by Metadata (
7dbbc99)
+Formatting changes to Dotplot by Metadata (
d994b6c)
+Code changes to Name clusters and formatting (
70a9fc2)
+Code changes to Name clusters and formatting (
bca98aa)
+Formatting changes to Heatmap.R (
e1bd5c7)
+Formatting changes to Heatmap.R (
f47e847)
+Add files via upload
+
replace image file (7bf9b80)
- Add files via upload +
replace image file (542b65d)
- Update README.md +
additional text (700ad07)
- Update README.md +
additional text (2e0feec)
- Update README.md +
Added image. (7694f8d)
- Update README.md +
Added image. (f28a6a7)
- Add files via upload +
Workflow image (f93175b)
- Add files via upload +
Workflow image (433a89d)
Create README.md (
70aa926)
+Create README.md (
0b318e5)
+formatting for dual labeling (
2e7d12c)
+formatting for dual labeling (
f782199)
+format 3D-tSNE function (
fe97cb0)
+format 3D-tSNE function (
4ae1381)
+Update gitflow-R-action.yml (
9bdcf6c)
+Update gitflow-R-action.yml (
7799784)
+update Dotplot conflict (
0ea5f42)
+update Dotplot conflict (
e99ac9b)
+Update (
aa71a29)
+Update (
3dfcdb5)
+Adding action files and dockerfiles (
d215fdb)
+Adding action files and dockerfiles (
1cbab8e)
+Resolving git conflict in Recluster. (
0c70bdb)
+Resolving git conflict in Recluster. (
28d7b96)
+committing in-progress Sugarloaf updates to Recluster SO template. (
ed03d46)
+committing in-progress Sugarloaf updates to Recluster SO template. (
446af26)
+changed fixture filenames (
71ebc2f)
+changed fixture filenames (
6e3284c)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
f5590de)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
219746d)
+edit code formatting (
1a7a120)
+edit code formatting (
2676adc)
+Initial TestThat for Recluster
+
Includes tests on all 5 datasets testing type, as well as one test on MouseTEC that tests UMAP instead of default TSNE. (4da52a7)
- Initial TestThat for Recluster +
Includes tests on all 5 datasets testing type, as well as one test on MouseTEC that tests UMAP instead of default TSNE. (da7c560)
Initial R4 functionalization. (
7558379)
+Initial R4 functionalization. (
84acb8c)
+Add files via upload
+
Initial release (3eca944)
- Add files via upload +
Initial release (deafd1c)
- Add files via upload +
Initial release (77b96a6)
- Add files via upload +
Initial release (91b7594)
- Add files via upload +
Initial commit (4a74c89)
- Add files via upload +
Initial commit (9d694ae)
- Add files via upload +
Initial release (fa2e7c3)
- Add files via upload +
Initial release (25d5857)
- Add files via upload +
Initial release (69d1dcf)
- Add files via upload +
Initial release (900ab4c)
Delete DEG_Gene_Expression_Markers.R (
5496f85)
+Delete DEG_Gene_Expression_Markers.R (
fe406ea)
+Add files via upload
+
Initial release (ea9b264)
- Add files via upload +
Initial release (45fd8d2)
- Add files via upload +
Initial release (c2e8e8c)
- Add files via upload +
Initial release (fee817b)
- Add files via upload +
Initial release (55fd6c9)
- Add files via upload +
Initial release (a59a2bc)
- Add files via upload +
Initial release (ad72a95)
- Add files via upload +
Initial release (aac8353)
- Add files via upload +
Initial release (a0c5606)
- Add files via upload +
Initial release (c31a9f2)
Delete DeletMeAgain (
d2436b6)
+Delete DeletMeAgain (
4514dae)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
9ce4628)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
2a53ebc)
+CommLine Test (
82aa0fa)
+CommLine Test (
811846c)
+Delete Delete.Me
+
Following the orders (6b41c15)
- Delete Delete.Me +
Following the orders (1f82ca7)
- Add files via upload +
DELETE IMMEDIATELY!!! (986326c)
- Add files via upload +
DELETE IMMEDIATELY!!! (ea68f0a)
Initial release (
2c12ea5)
+Initial release (
a294bd3)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
a276a80)
+Recover code from past commit (
184c917)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
update function and parameter names. (83fbdd1)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
update function and parameter names. (6f46e6e)
reformat function and parameter names (
0dfca6c)
+reformat function and parameter names (
c81712f)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
eac38ce)
+Merge pull request #9 from NIDAP-Community/Rui_resolve_conflict
+
Resolve conflicts (7fa7db9)
- Merge pull request #9 from NIDAP-Community/Rui_resolve_conflict +
Resolve conflicts (7839aba)
Resolve conflicts (
d516cd2)
+Resolve conflicts (
8c776b1)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
0a5942a)
+Update main on local branch (
b27d7af)
+Merge pull request #8 from NIDAP-Community/phi_test
+
Phi test (2d5e7ec)
- Merge pull request #8 from NIDAP-Community/phi_test +
Phi test (87fdb1a)
resolve conflict (
b4d8548)
+resolve conflict (
a3ddae1)
+Merge branch 'main' into phi_test (
e22b5e6)
+Add ignore h5 files in gitignore (
55c3167)
+resolve conflicts (
43976ba)
+resolve error (
d59173b)
+Update current directory (
f7e242b)
+Downsampled CITEseq (
bdfa1f3)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main Correct CITEseq Downsmple (
4d643f4)
+correct CITEseq Downsample (
3f032fd)
+helper script for 3D-tsne (
12ee9f3)
+helper script for 3D-tsne (
30b7146)
+helper script for 3D-tsne (
a901830)
+new tests (
1ce5664)
+new tests (
29d75bc)
+new tests (
9cb29d2)
+unit test Jing templates (
bb48481)
+unit test Jing templates (
63a6636)
+unit test Jing templates (
97d74ca)
+NSCLCmulti SO (
3d42d4a)
+NSCLCmulti SO (
540bdf3)
+NSCLCmulti SO (
6e8e596)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
Update Chariou Single R SO and BRCA combin and Renormalize (e851077)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
Update Chariou Single R SO and BRCA combin and Renormalize (072096f)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
Update Chariou Single R SO and BRCA combin and Renormalize (1a8fbf8)
BRCA comb_Renorm (
5e3985b)
+BRCA comb_Renorm (
9fb63a3)
+BRCA comb_Renorm (
6d4c05a)
+unit tests for Name Clusters (
95cf6f5)
+unit tests for Name Clusters (
c7ed110)
+unit tests for Name Clusters (
43859bd)
+unit test for dual labeling (
46dd45e)
+unit test for dual labeling (
3f07269)
+unit test for dual labeling (
5c85127)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
merging new changes (b8b1eda)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
merging new changes (09c8f46)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
merging new changes (729b051)
unit tests for heatmap, dotplot (
7659b83)
+unit tests for heatmap, dotplot (
b81287c)
+unit tests for heatmap, dotplot (
5414455)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
adding NSCLC_Single SOs (152cf11)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
adding NSCLC_Single SOs (b0ea506)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
adding NSCLC_Single SOs (d83c561)
NSCLC_single SO (
df265cd)
+NSCLC_single SO (
ad62924)
+NSCLC_single SO (
7d0b242)
+add dotplot tests (
57166ba)
+add dotplot tests (
b2bd01e)
+add dotplot tests (
69fec28)
+unit test for Dotplot (
a94f7ba)
+unit test for Dotplot (
13fb938)
+unit test for Dotplot (
9c67e81)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
new error messaging added (b09cb64)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
new error messaging added (d266d5e)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
new error messaging added (c0aaa1b)
new error messaging to dotplot (
9742c5f)
+new error messaging to dotplot (
a9bf495)
+new error messaging to dotplot (
93a3a06)
+changes to dotplot (
1849346)
+changes to dotplot (
b52dc29)
+changes to dotplot (
d4c0824)
+Charou SO (
dccaf54)
+Charou SO (
95b3421)
+Charou SO (
1f3ca66)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main
+
Conflicts: .gitignore DESCRIPTION tests/testthat/test_Filter_and_QC.R (c58b92e)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
Conflicts: .gitignore DESCRIPTION tests/testthat/test_Filter_and_QC.R (8a845be)
- Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main +
Conflicts: .gitignore DESCRIPTION tests/testthat/test_Filter_and_QC.R (bb18f4c)
The conda package build requires a NAMESPACE file to be added, which is being ignored during the dev cycle. (e7ac125)
- NAMESPACE removed from merge +
The conda package build requires a NAMESPACE file to be added, which is being ignored during the dev cycle. (0027492)
- NAMESPACE removed from merge +
The conda package build requires a NAMESPACE file to be added, which is being ignored during the dev cycle. (4f3bbc8)
- Merge pull request #7 from NIDAP-Community/dev_release_dec_13_22 +
Dev release dec 13 22 (02999db)
- Merge pull request #7 from NIDAP-Community/dev_release_dec_13_22 +
Dev release dec 13 22 (6ec32fa)
- Merge pull request #7 from NIDAP-Community/dev_release_dec_13_22 +
Dev release dec 13 22 (2e0f434)
Initial Commit for sprint 5 Functions. Including change to DESCRIPTION file (
4384dd1)
+Initial Commit for sprint 5 Functions. Including change to DESCRIPTION file (
eeb7741)
+Initial Commit for sprint 5 Functions. Including change to DESCRIPTION file (
90120cb)
+Minor fix for codes to pass Check (
3b4f7ee)
+Minor fix for codes to pass Check (
29de7d6)
+Minor fix for codes to pass Check (
b65e314)
+Updated tests (
733e2ec)
+Updated tests (
c9ed47b)
+Updated tests (
97e18e5)
+update NameClusters function and test (
128fbb4)
+update NameClusters function and test (
3f90257)
+update NameClusters function and test (
c88161e)
+update test-Metadata_Table.R (
a37474b)
+update test-Metadata_Table.R (
4257cc6)
+update test-Metadata_Table.R (
7b2f599)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
78fc249)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
cce3673)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
986e82b)
+small changes in 3D plotter testing (
74364db)
+small changes in 3D plotter testing (
a60aa38)
+small changes in 3D plotter testing (
7ac6a75)
+changed heatmap test and added library to heatmap (
cb33cf2)
+changed heatmap test and added library to heatmap (
bcab3c2)
+changed heatmap test and added library to heatmap (
20f7260)
+revised test for dual labeling (
b5caf02)
+revised test for dual labeling (
0f39220)
+revised test for dual labeling (
d1b47d6)
+new doc for dotplot (
b986ccc)
+new doc for dotplot (
800edc4)
+new doc for dotplot (
acd4514)
+changes to test and added color option (
493b745)
+changes to test and added color option (
b66d7fa)
+changes to test and added color option (
69c3c15)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
8863d0d)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
63fc3f6)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
8fa25f6)
+add original citation for Pseudobulk.R (
4293f96)
+add original citation for Pseudobulk.R (
53add2e)
+add original citation for Pseudobulk.R (
7b99980)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
9abcb98)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
940e610)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
fb13a18)
+add functions for heatmap, dotplot, 3d-tsne and dual-labeling (
0ed4df7)
+add functions for heatmap, dotplot, 3d-tsne and dual-labeling (
74fe2ad)
+add functions for heatmap, dotplot, 3d-tsne and dual-labeling (
44338d2)
+update ModScore Pseudobulk and DESCRIPTION (
aefedde)
+update ModScore Pseudobulk and DESCRIPTION (
abe0919)
+update ModScore Pseudobulk and DESCRIPTION (
ec95f0c)
+fix NAMESPACE @importFrom methods empty (
756de4e)
+fix NAMESPACE @importFrom methods empty (
2f2bd5a)
+fix NAMESPACE @importFrom methods empty (
5984060)
+Update Description (
3dcc872)
+Update Description (
968b204)
+Update Description (
1f05120)
+Merge pull request #6 from NIDAP-Community/rui
+
Rui (c09fa3f)
- Merge pull request #6 from NIDAP-Community/rui +
Rui (5d3d3b2)
- Merge pull request #6 from NIDAP-Community/rui +
Rui (e71621d)
update man docs (
bf9fb25)
+update man docs (
c08890f)
+update man docs (
98e86e3)
+add NameCluster function and tests (
dbe4d15)
+add NameCluster function and tests (
5812a98)
+add NameCluster function and tests (
233dd75)
+update MetadataTable & SampleNames (
b3f19d8)
+update MetadataTable & SampleNames (
175fa43)
+update MetadataTable & SampleNames (
1d921a2)
+drop unused factor levels in SO_moduleScore.rds (
3a39fcf)
+drop unused factor levels in SO_moduleScore.rds (
133d872)
+drop unused factor levels in SO_moduleScore.rds (
ea6df85)
+Jing templates and fixtures (
08532bf)
+Jing templates and fixtures (
9f84531)
+Jing templates and fixtures (
150df39)
+add SampleNames .R and .Rd files (
9f16588)
+add SampleNames .R and .Rd files (
780b298)
+add SampleNames .R and .Rd files (
446d631)
+update MetadataTable (
615afdc)
+update MetadataTable (
44abfce)
+update MetadataTable (
a27296a)
+Unit test added to 3d tsne function (
3ab8d75)
+Unit test added to 3d tsne function (
6dc17bd)
+Unit test added to 3d tsne function (
e032618)
+rui update 1 (
9011356)
+rui update 1 (
81793d8)
+rui update 1 (
7006ac8)
+add man/MetadataTable.Rd (
af8eedf)
+add man/MetadataTable.Rd (
7830b05)
+add man/MetadataTable.Rd (
61457e8)
+upload fixtures/SO_moduleScore.rds (
78caf56)
+upload fixtures/SO_moduleScore.rds (
07bc42d)
+upload fixtures/SO_moduleScore.rds (
b3b3dc4)
+correct DESCRIPTION tibble (
57847f5)
+correct DESCRIPTION tibble (
fe89882)
+correct DESCRIPTION tibble (
90dd394)
+update DESCRIPTION (
f55cf25)
+update DESCRIPTION (
fcf6d26)
+update DESCRIPTION (
7d3e4ef)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
be5cff8)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
0d74815)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
58aa02a)
+add MetadataTable function (
42c3569)
+add MetadataTable function (
50e3af0)
+add MetadataTable function (
09b995d)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
01921d5)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
795043f)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
70082b0)
+removed NAMESPACE from repo (
dc422fa)
+removed NAMESPACE from repo (
d528f76)
+removed NAMESPACE from repo (
f7f5da5)
+removed Rcheck.txt (
8b70481)
+removed Rcheck.txt (
e0a6384)
+removed Rcheck.txt (
e23003a)
+Added Git ignore and update R check results, 11_16_2022 (
ce18e14)
+Added Git ignore and update R check results, 11_16_2022 (
e705893)
+Added Git ignore and update R check results, 11_16_2022 (
1d8a95c)
+Update DESCRIPTION (
7fdea8e)
+Update DESCRIPTION (
0d266f4)
+Update DESCRIPTION (
5effd23)
+Update cc.genes calls (
797aca1)
+Update cc.genes calls (
3db02b2)
+Update cc.genes calls (
65f805b)
+Update cc.genes calls (
8cd8b03)
+Update cc.genes calls (
82a4244)
+Update cc.genes calls (
ea8e671)
+Update library call (
423fbe9)
+Update library call (
f52a89c)
+Update library call (
a3d302b)
+Update namespace (
a4230b7)
+Update namespace (
d35dc0e)
+Update namespace (
0a8e2a9)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
272c0ec)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
8558d4f)
+Merge branch 'main' of https://github.com/NIDAP-Community/SCWorkflow into main (
83f6f23)
+Update namespace (
9960214)
+Update namespace (
3d85b8d)
+Update namespace (
b8c8b0e)
+Merge pull request #5 from NIDAP-Community/initial_filter_qc
+
Adjusted png render method for NIDAP display (33820ce)
- Merge pull request #5 from NIDAP-Community/initial_filter_qc +
Adjusted png render method for NIDAP display (770cb6a)
- Merge pull request #5 from NIDAP-Community/initial_filter_qc +
Adjusted png render method for NIDAP display (12b4ce3)
Adjusted png render method for NIDAP display (
0b9e4d2)
+Adjusted png render method for NIDAP display (
9968f1f)
+Adjusted png render method for NIDAP display (
747b89a)
+Merge pull request #4 from NIDAP-Community/initial_filter_qc
+
Update namespace (bcc1c7f)
- Merge pull request #4 from NIDAP-Community/initial_filter_qc +
Update namespace (61952ec)
- Merge pull request #4 from NIDAP-Community/initial_filter_qc +
Update namespace (ac05afc)
Update namespace (
fdcfd52)
+Update namespace (
f070d8e)
+Update namespace (
fd74f19)
+Merge pull request #3 from NIDAP-Community/initial_filter_qc
+
Added license file (a3a99b6)
- Merge pull request #3 from NIDAP-Community/initial_filter_qc +
Added license file (5c508c3)
- Merge pull request #3 from NIDAP-Community/initial_filter_qc +
Added license file (d5113f3)
Added license file (
186d183)
+Added license file (
a8bd74c)
+Added license file (
1d7ed13)
+Merge pull request #2 from NIDAP-Community/initial_filter_qc
+
remove GenomeInfoDb (fe8164b)
- Merge pull request #2 from NIDAP-Community/initial_filter_qc +
remove GenomeInfoDb (75ae5fd)
- Merge pull request #2 from NIDAP-Community/initial_filter_qc +
remove GenomeInfoDb (8908e24)
remove GenomeInfoDb (
f3bb0e6)
+remove GenomeInfoDb (
2b32d4e)
+remove GenomeInfoDb (
b27279e)
+Merge pull request #1 from NIDAP-Community/initial_filter_qc
+
Initial push with filter and qc, demo (866a648)
- Merge pull request #1 from NIDAP-Community/initial_filter_qc +
Initial push with filter and qc, demo (a30130b)
- Merge pull request #1 from NIDAP-Community/initial_filter_qc +
Initial push with filter and qc, demo (b627f06)
Initial push with filter and qc, demo (
a843d4d)
+Initial push with filter and qc, demo (
dcd9fef)
+Initial push with filter and qc, demo (
462a39d)
+Update README.md
+
Changed package name (acb154c)
- Update README.md +
Changed package name (fdffdf4)
+
+
+
+
+
+
+
+
diff --git a/docs/LICENSE.html b/docs/LICENSE.html
new file mode 100644
index 0000000..61736ff
--- /dev/null
+++ b/docs/LICENSE.html
@@ -0,0 +1,71 @@
+
+
+
+
+
+
+
+
+
+License
+ +YEAR: 2024 +COPYRIGHT HOLDER: NIDAP Community ++ +
+
+
+
+
+
+
+
+
diff --git a/docs/articles/CONTRIBUTING.html b/docs/articles/CONTRIBUTING.html
new file mode 100644
index 0000000..31a76ec
--- /dev/null
+++ b/docs/articles/CONTRIBUTING.html
@@ -0,0 +1,526 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+NA
+ +MIT License
+Copyright (c) 2024 NIDAP Community
+Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the “Software”), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
+The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
+THE SOFTWARE IS PROVIDED “AS IS”, WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
+ + +
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/Intro.html b/docs/articles/Intro.html
new file mode 100644
index 0000000..91ada33
--- /dev/null
+++ b/docs/articles/Intro.html
@@ -0,0 +1,167 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Contributing to SCWorkflow
+ + + +CONTRIBUTING.Rmd
+
+
++Propose Change +
++
+
++Clone the repo +
+If you are a member of CCBR, +you can clone this repository to your computer or development +environment.
+SCWorkflow is a large repository so this may take a few minutes.
+ +++ +Cloning into ‘SCWorkflow’…
+
remote: Enumerating objects: 3126, +done.
remote: Counting objects: 100% (734/734), done.
remote: +Compressing objects: 100% (191/191), done.
remote: Total 3126 +(delta 630), reused 545 (delta 543), pack-reused 2392 (from 1)
+Receiving objects: 100% (3126/3126), 1.04 GiB | 4.99 MiB/s, done.
+Resolving deltas: 100% (1754/1754), done.
Updating files: 100% +(306/306), done.
+
+
++Install dependencies +
+If this is your first time cloning the repo you may have to install +dependencies
+ +Check R CMD: In an R console, make sure the package +passes R CMD check by running:
+
+ devtools::check()++ +⚠️ Note: If R CMD check doesn’t pass cleanly, it’s a +good idea to ask for help before continuing.
+
+
+
++Load SCWorkflow from repo +
+In an R console, load the package from the local repo using:
+
+devtools::load_all()
+
+
++Create branch +
+Create a Git branch for your pull request (PR). Give the branch a +descriptive name for the changes you will make.
+Example: Use iss-10 if it’s for a
+specific issue, or feature-new-plot for a new feature.
For bug fixes or small changes, you can branch from the
+main branch.
++Success: Switched to a new branch ‘iss-10’
+
For new features or larger changes, branch from the DEV
+branch.
# Switch to DEV branch, create a new branch, and switch to new branch
+git switch DEV
+git branch feature-new-plot
+git switch feature-new-plot++Success: Switched to a new branch +‘feature-new-plot’
+
+
+
+Develop +
++
+
++Make your changes +
+Now you’re ready to edit the code, write unit tests, and update the +documentation as needed.
+
+
+
+
++Code Style Guidelines +
+New code should follow the general guidelines outlined here. +- Important: Don’t restyle code unrelated to your +PR
+Tools to help: - Use the styler package to +apply these styles
+Key conventions from the tidyverse style +guide:
+| Element | +Style | +Example | +
|---|---|---|
| Variables | +snake_case | +my_variable |
+
| Functions | +verbs in camelCase | +processData() |
+
| Assignment | +
+<- operator |
+x <- 5 |
+
| Operations | +pipes | +data %>% filter() %>% mutate() |
+
+
+
++Function Organization +
+Structure your functions like this:
+Functions should follow this template. Use roxygen2 for +documentation:
+
+#' @title Function Title
+#' @description Brief description of what the function does
+#' @param param1 Description of first parameter
+#' @param param2 Description of second parameter
+#' @details Additional details if needed
+#' @importFrom package function_name
+#' @export
+#' @return Description of what the function returns
+
+yourFunction <- function(param1, param2) {
+
+ ## --------- ##
+ ## Functions ##
+ ## --------- ##
+
+ ## --------------- ##
+ ## Main Code Block ##
+ ## --------------- ##
+
+ output_list <- list(
+ object = SeuratObject,
+ plots = list(
+ 'plotTitle1' = p1,
+ 'plotTitle2' = p2
+ ),
+ data = list(
+ 'dataframeTitle' = df1
+ )
+ )
+
+ return(output_list)
+}
+
++Commit and Push Your Changes +
+Best practices for commits:
+We recommend following the “atomic commits” +principle where each commit contains one new feature, fix, or task.
+Learn more: Atomic +Commits Guide
+
+
++Step-by-Step Process: +
+ + +
+
+3️⃣ Make the Commit +
+ +Your commit message should follow the Conventional
+Commits specification. Briefly, each commit should start with one of
+the approved types such as feat, fix,
+docs, etc. followed by a description of the commit. Take a
+look at the Conventional
+Commits specification for more detailed information about how to
+write commit messages.
+
+4️⃣ Push your changes to GitHub: +
+ +If this is the first time you are pushing this branch, you may have +to explicitly set the upstream branch:
+ +We recommend pushing your commits often so they will be backed up on
+GitHub. You can view the files in your branch on GitHub at
+https://github.com/NIDAP-Community/SCWorkflow/tree/<your-branch-name>
+(replace <your-branch-name> with the actual name of
+your branch).
+
+
+Document and Tests +
++
+
++Writing Tests +
+Why tests matter: Most changes to the code will also +need unit tests to demonstrate that the changes work as intended.
+How to add tests:
+-
+
- Use
testthat+to create your unit tests
+ - Follow the organization described in the tidyverse test style +guide + +
- Look at existing code in this package for examples +
+
++Documentation +
+When to update documentation:
+-
+
- Written a new function +
- Changed the API of an existing function +
- Function is used in a vignette +
How to update documentation:
+-
+
- Use roxygen2 with Markdown +syntax + +
- See the R Packages book +for detailed instructions +
- Update relevant vignettes if needed +
+
+
++Check Your Work +
+🔍 Final validation step:
+After making your changes, run the following command from an R +console to make sure the package still passes R CMD check:
+
+devtools::check()++Goal: All checks should pass with no errors, +warnings, or notes.
+
+
+
++Deploy Feature +
++
+
+1️⃣ Create the PR +
+Once your branch is ready, create a PR on GitHub: https://github.com/NIDAP-Community/SCWorkflow/pull/new/
+Select the branch you just pushed:
+
+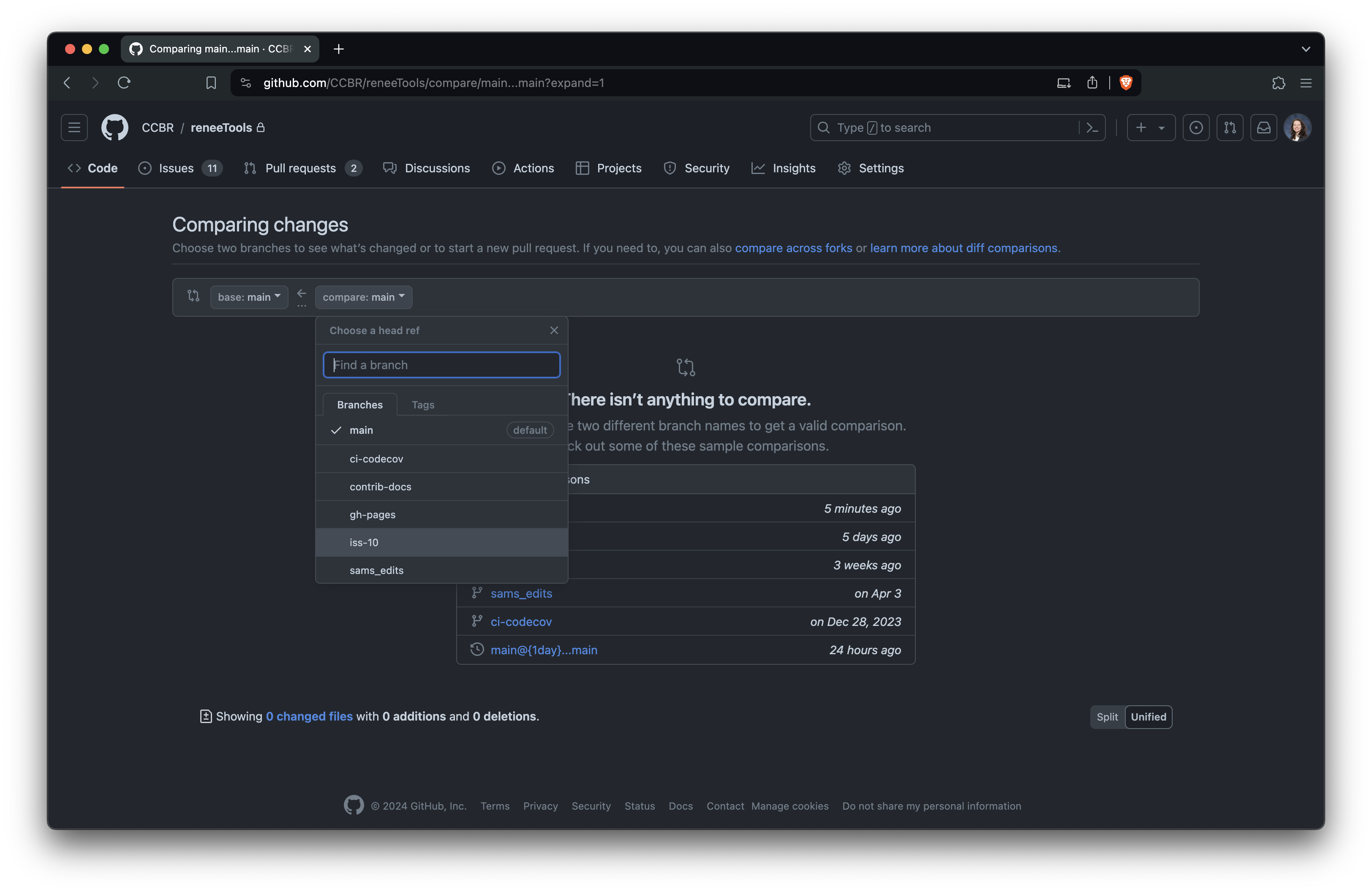
+Create a new PR from your branch
+Edit the PR title and description. The title should briefly describe
+the change. Follow the comments in the template to fill out the body of
+the PR, and you can delete the comments (everything between
+<!-- and -->) as you go. When you’re
+ready, click ‘Create pull request’ to open it.
+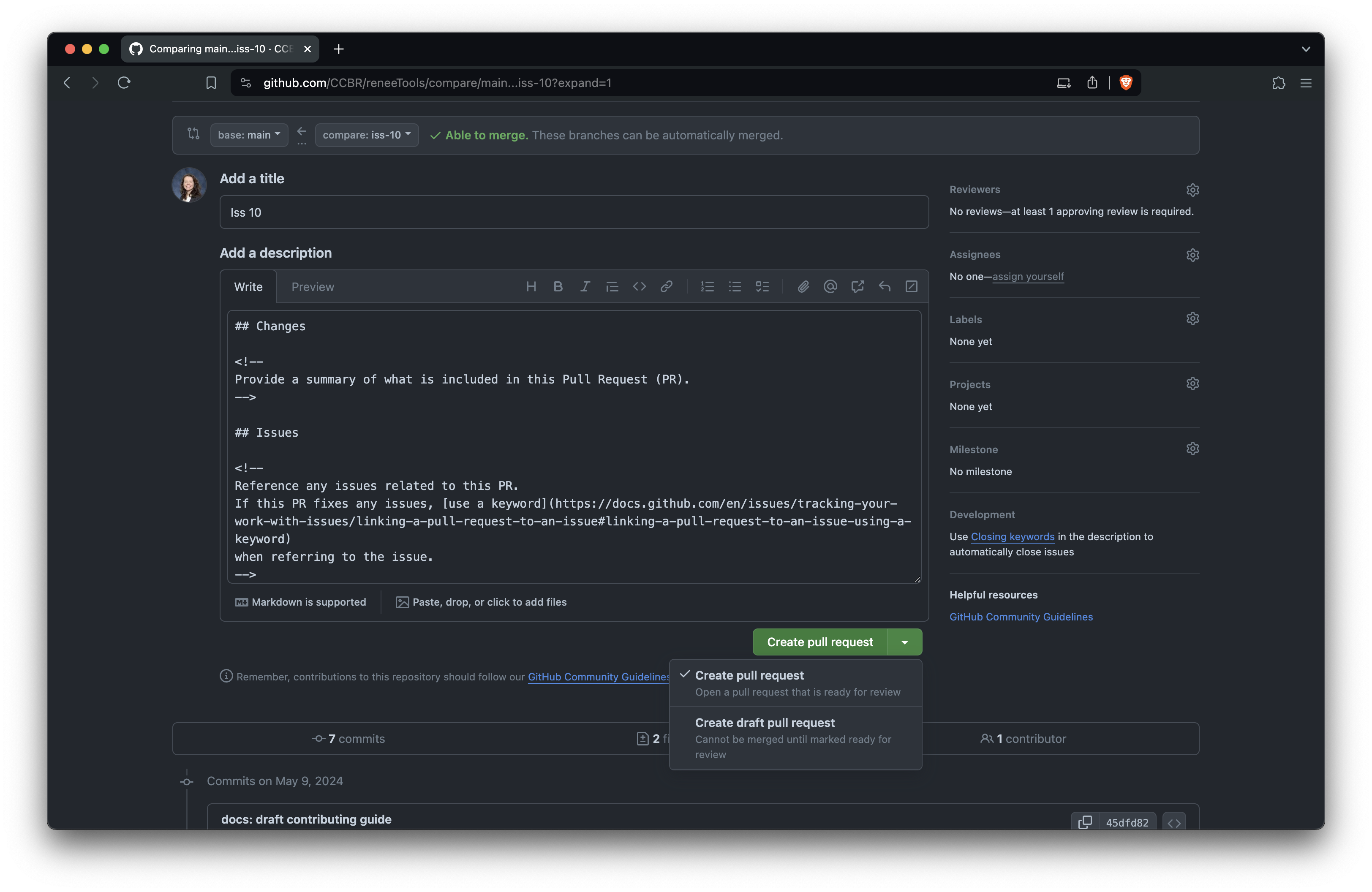
+Open the PR after editing the title and
+description
+Optionally, you can mark the PR as a draft if you’re not yet ready +for it to be reviewed, then change it later when you’re ready.
+
+
+
+2️⃣ Wait for a maintainer to review your PR +
+We will do our best to follow the tidyverse code review principles: +https://code-review.tidyverse.org/. The reviewer may +suggest that you make changes before accepting your PR in order to +improve the code quality or style. If that’s the case, continue to make +changes in your branch and push them to GitHub, and they will appear in +the PR.
+Once the PR is approved, the maintainer will merge it and the +issue(s) the PR links will close automatically. Congratulations and +thank you for your contribution!
+
+
+ +Helpful links for more information +
+-
+
- This contributing guide was adapted from the tidyverse +contributing guide + +
- GitHub +Flow +
- tidyverse style guide +
- tidyverse code review +principles +
- reproducible +examples +
- R packages book +
- packages: + + +
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/README.html b/docs/articles/README.html
new file mode 100644
index 0000000..d1bc74f
--- /dev/null
+++ b/docs/articles/README.html
@@ -0,0 +1,167 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Intro.Rmd
+
+ SCWorkflow +
+The CCBR Single-cell RNA-seq Package (SCWorkflow) allows users to +analyze their own single-cell RNA-seq datasets starting from CellRanger +output files (H5 or mtx files, etc.).
+
+
+
+
+
+Installation +
++
You can install the SCWorkflow package from GitHub +with:
+
+# install.packages("remotes")
+remotes::install_github("NIDAP-Community/SCWorkflow", dependencies = TRUE)There is also a Docker container available at
+
+
+
+Usage +
++
Following this workflow you can perform these steps of a single-cell +RNA-seq analysis, and more:
+-
+
-
+
Quality Control:
+-
+
Import, Select, & Rename Samples
+Filter Cells based on QC metrics
+Combine Samples, Cluster, and Normalize your Data
+Batch Correction using Harmony
+
+ -
+
Cell Annotation:
+-
+
SingleR Automated Annotations
+Module Scores
+Co-Expression
+External Annotations
+
+ -
+
Visualizations:
+-
+
Dimensionality Reductions (t-SNE and UMAP Plots) colored by +Marker Expression or by Metadata
+Heatmaps
+Violin Plots
+Trajectory
+
+ -
+
Differential Expression Analysis
+-
+
Seurat’s FindMarkers()
+Pseudobulk Aggregation
+Pathway Analysis
+
+
Please see the introductory +vignette for a quick start tutorial. Take a look at the reference +documentation for detailed information on each function in the +package.
+
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/SCWorkflow-Annotations.html b/docs/articles/SCWorkflow-Annotations.html
new file mode 100644
index 0000000..3feac25
--- /dev/null
+++ b/docs/articles/SCWorkflow-Annotations.html
@@ -0,0 +1,450 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+SCWorkflow-Intro
+ + + +README.Rmd
+
+ SCWorkflow +
+The CCBR Single-cell RNA-seq Package (SCWorkflow) allows users to +analyze their own single-cell RNA-seq datasets starting from CellRanger +output files (H5 or mtx files, etc.).
+
+
+
+
+
+Installation +
++
You can install the SCWorkflow package from GitHub +with:
+
+# install.packages("remotes")
+remotes::install_github("NIDAP-Community/SCWorkflow", dependencies = TRUE)There is also a Docker container available at
+
+
+
+Usage +
++
Following this workflow you can perform these steps of a single-cell +RNA-seq analysis, and more:
+-
+
-
+
Quality Control:
+-
+
Import, Select, & Rename Samples
+Filter Cells based on QC metrics
+Combine Samples, Cluster, and Normalize your Data
+Batch Correction using Harmony
+
+ -
+
Cell Annotation:
+-
+
SingleR Automated Annotations
+Module Scores
+Co-Expression
+External Annotations
+
+ -
+
Visualizations:
+-
+
Dimensionality Reductions (t-SNE and UMAP Plots) colored by +Marker Expression or by Metadata
+Heatmaps
+Violin Plots
+Trajectory
+
+ -
+
Differential Expression Analysis
+-
+
Seurat’s FindMarkers()
+Pseudobulk Aggregation
+Pathway Analysis
+
+
Please see the introductory +vignette for a quick start tutorial. Take a look at the reference +documentation for detailed information on each function in the +package.
+
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/SCWorkflow-DEG.html b/docs/articles/SCWorkflow-DEG.html
new file mode 100644
index 0000000..2d6a8fc
--- /dev/null
+++ b/docs/articles/SCWorkflow-DEG.html
@@ -0,0 +1,501 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Annotations
+ + + +SCWorkflow-Annotations.Rmd
+
+
+
+Cell Type Annotation with SingleR +
++
This function automates cell type annotation in single-cell RNA +sequencing data by employing the SingleR [1] method, which +allocates labels to cells within a dataset according to their gene +expression profile similarities with a reference dataset containing cell +type labeled samples
+SingleR is an automatic annotation method for single-cell RNA +sequencing data that uses a given reference dataset of samples +(single-cell or bulk) with known labels to label new cells from a test +dataset based on similarity to the reference. Two mouse reference +datasets (MouseRNAseqData and ImmGenData) and two human reference +datasets (HumanPrimaryCellAtlasData and BlueprintEncodeData) from +CellDex R package [2] are currently available.
+
+
+Anno_SO=annotateCellTypes(object=Comb_SO$object,
+ species = "Mouse",
+ reduction.type = "umap",
+ legend.dot.size = 2,
+ do.finetuning = FALSE,
+ local.celldex = NULL,
+ use.clusters = NULL
+ )

+
+
+
+Cell Annotation with Co-Expression +
++
This function will display co-expression of two chosen markers in +your Seurat Object. It will then Create a metadata column containing +annotations for cells that correspond to the marker expression +thresholds you set.
+This function enables users to visualize the association between two +selected genes or proteins according to their expression values in +various samples. The association is plotted, and samples with values +above or below a specified limit can be excluded. Users have the ability +to customize the visualization, including the choice of visualization +type, point size and shape, and transparency level.
+Furthermore, the tool allows for the application of filters to the +data, setting thresholds, and providing annotations to notify users if +cells meet the established thresholds. The visualization can be improved +by omitting extreme values. The tool also facilitates the creation of a +heatmap to represent the density distribution of cells and exhibit the +raw gene/protein expression values.
+
+grep("Cd4",rownames(Anno_SO$object@assays$RNA),ignore.case = T,value=T)
+
+DLAnno_SO=dualLabeling(object = Anno_SO$object,
+ samples <- c("PBS","CD8dep","ENT","NHSIL12","Combo"),
+ marker.1="Nos2",
+ marker.2="Arg1",
+ marker.1.type = "SCT",
+ marker.2.type = "SCT",
+ data.reduction = "both",
+ point.size = 0.5,
+ point.shape = 16,
+ point.transparency = 0.5,
+ add.marker.thresholds = TRUE,
+ marker.1.threshold = 0.5,
+ marker.2.threshold = 0.5,
+ filter.data = TRUE,
+ marker.1.filter.direction = "greater than",
+ marker.2.filter.direction = "greater than",
+ apply.filter.1 = TRUE,
+ apply.filter.2 = TRUE,
+ filter.condition = TRUE,
+ parameter.name = "My_CoExp",
+ trim.marker.1 = FALSE,
+ trim.marker.2 = FALSE,
+ pre.scale.trim = 0.99,
+ display.unscaled.values = FALSE
+ )
+
+plot(DLAnno_SO$plots$tsne)
+plot(DLAnno_SO$plots$umap)

+
+
+
+
+Color by Gene Lists +
++
This function generates plots to visualize the expression of specific +markers (genes) in single-cell RNA sequencing (scRNA-seq) data. Gene +plots are generated in the same order as they appear in the input list +(provided that they are present in the data).
+This function takes in a number of inputs to create detailed plots +showing the activity of certain genes. You can customize these based on +specific samples or genes of interest or point transparency. The code +has a built-in system to alert you if there are any issues with your +chosen inputs. If a particular gene is missing, it will return an empty +plot. If the gene is present, it will perform several steps to adjust +the data for better visualization and analysis, such as normalizing the +data and creating a reduction plot (a type of plot that helps visualize +complex data). The code also makes sure to display your chosen samples, +creates a caption for the plot indicating which samples are shown, +colors the points based on gene activity levels, and adjusts the plot’s +visual elements like transparency, size, and labels. If you haven’t +selected specific samples, it will use all the available samples from +your data. It also checks for the presence of your chosen genes in the +data and processes them to ensure uniformity across different cell +types. The output of this function is a detailed figure showing the +activity of chosen genes across different cell types. This is useful for +identifying distinct groups of cells based on gene activity levels.
+
+Marker_Table <- read.csv("Marker_Table_demo.csv")
+
+ colorByMarkerTable(object=Anno_SO$object,
+ samples.subset=c("PBS","ENT","NHSIL12", "Combo","CD8dep" ),
+ samples.to.display=c("PBS","ENT","NHSIL12", "Combo","CD8dep" ),
+ marker.table=Marker_Table,
+ cells.of.interest=c("Neutrophils","Macrophages","CD8_T" ),
+ protein.presence = FALSE,
+ assay = "SCT",
+ reduction.type = "umap",
+ point.transparency = 0.5,
+ point.shape = 16,
+ cite.seq = FALSE
+ )
+
+
+
+Module Score Cell Classification +
++
Screens data for cells based on user-specified cell markers. Outputs +a seurat object with a metadata with averaged marker scores and +annotated “Likely_CellType” column.
+This function can be used to quantify the expression of marker sets +in each individual cell and (optionally) in a hierarchical manner, +helping you identify different cell types and potential +subpopulations.
+This function aids in identifying cell types based on average gene +expression. It uses a feature of the Seurat software known as the +AddModuleScore function. This function calculates the gene expression of +specific sets and records them within a designated area of the Seurat +object. The program then forecasts cell identities by comparing these +recorded scores across various gene sets. You have the ability to adjust +the identification process by designating cutoff points for a bimodal +distribution in a parameter known as manual threshold. Any thresholds +below this cutoff will not be considered during the identification +process.
+Inputs: The program takes several inputs. These +include the single-cell RNA sequencing (scRNA-seq) object, a selection +of samples for analysis, a table of gene markers for different cell +types, and optionally, a hierarchical table for directing the order of +cell classification. Data Preparation: The program +prepares the scRNA-seq object, assigns names to the samples, and selects +data based on your specified samples. Module Score +Calculation: The program calculates module scores, a measure of +gene set activity or expression [3], for each cell type based on your +provided marker table. Visualization: Density +distribution plots and colored reduction plots will be generated to help +you visualize the module scores, their relationship with cell types, and +sample identities. Thresholding: Users can select +threshold values to aid in the classification of cells. Cells with +scores below your designated threshold will be labeled as “unknown”. +Subclass Identification: If desired, the program can identify subclasses +within cell types by further analyzing subpopulations. Updating +Cell Type Labels: The program appends a “Likely_CellType” +column to the metadata of the scRNA-seq object, based on the results of +the module score analysis. Output: An updated scRNA-seq +object with new cell type labels.
+
+
+MS_object=modScore(object=Anno_SO$object,
+ marker.table=Marker_Table,
+ use.columns = c("Neutrophils","Macrophages","CD8_T" ),
+ ms.threshold=c("Neutrophils .25","Macrophages .40","CD8_T .14"),
+ general.class=c("Neutrophils","Macrophages","CD8_T"),
+ multi.lvl = FALSE,
+ reduction = "umap",
+ nbins = 10,
+ gradient.ft.size = 6,
+ violin.ft.size = 6,
+ step.size = 0.1
+ )


+
+
+
+
+
+
+
+Rename Clusters by Cell Type +
++
This function creates a dot plot of Cell Types by Renamed Clusters +and outputs a Seurat Object with a new metadata column containing these +New Cluster Names. The Cell Types are often determined by looking at the +Module Score Cell Classification calls made by the upstream Module Score +Cell Classification (see MS_Celltype metadata column).
+You must provide a table with a column containing the unique Cluster +IDs from an upstream clustering analysis (e.g. one of the SCT_snn_res_* +columns in your input Seurat Object metadata) and a column containing +the corresponding New Cluster Names you have chosen. The dot plot will +display the unique Cell Types on the x-axis and the Renamed Clusters on +the y-axis. The size of the dots show the percentage of cells in each +row (each Renamed Cluster) that was classified as each Cell Type. A +comparison of dot sizes within a row may provide insights into that +cluster’s primary Cell Type. A new metadata column named “Clusternames” +is added to the output Seurat Object that contains the New Cluster +Names.
+Methodology
+This function creates a dot plot visualization of cell types by metadata
+category (usually cluster number) from a given dataset implemented in
+the SCWorkflow package. The function allows you to update and organize
+biological data about cell clusters in a Seurat object. By changing the
+input labels, you can map custom names to the existing cluster IDs which
+will add these names to a new metadata column. It also generates a dot
+plot using Seurat’s Dotplot function [4], providing a visual
+representation of the percentage of various cell types within each
+cluster. Typically, a cluster can be more distinctively named by the
+predominant cell type as seen in the dotplot. The plot’s order can be
+customized for the clusters and cell types. If no specific order is
+provided, the function uses a default order. An optional parameter
+allows the user to make the plot interactive. The function returns the
+updated Seurat object and the plot.
+
+clstrTable <- read.table(file = "./images/Cluster_Names.txt", sep = '\t',header = T)| OriginalClusterIDs | +NewClusterNames | +
|---|---|
| 0 | +Pop0 | +
| 1 | +Pop1 | +
| 2 | +Pop2 | +
| 3 | +Pop3 | +
| 4 | +Pop4 | +
| 5 | +Pop5 | +
| 6 | +Pop6 | +
| 7 | +Pop7 | +
+
+RNC_object=nameClusters(object=Anno_SO$object,
+ cluster.identities.table=clstrTable,
+ cluster.numbers= 'OriginalClusterIDs',
+ cluster.names='NewClusterNames',
+ cluster.column ="SCT_snn_res.0.2",
+ labels.column = "mouseRNAseq_main",
+ order.clusters.by = NULL,
+ order.celltypes.by = NULL,
+ interactive = FALSE
+ )
+
+# DimPlot(MS_object, group.by = "SCT_snn_res.0.2", label = T, reduction = 'umap')
+# DimPlot(MS_object, group.by = "mouseRNAseq_main", label = T, reduction = 'umap')
+
+ggsave(RNC_object$plots, filename = "./images/RNC.png", width = 9, height = 6)
+
+
+
+ Dot Plot of Genes by Metadata +
++
This function creates a dot plot of average gene expression values +for a set of genes in cell subpopulations defined by metadata annotation +columns. The input table contains a single column for genes (the “Genes +column”) and a single column for category (the “Category labels to plot” +column). The values in the “Category labels to plot” column should match +the values provided in the metadata function (Metadata Category to +Plot). The plot will order the genes (x-axis, left to right) and +Categories (y-axis, top to bottom) in the order in which it appears in +the input table. Any category entries omitted will not be plotted.
+The Dotplot size will reflect the percentage of cells expressing the +gene while the color will reflect the average expression for the gene. A +table showing values on the plot (either percentage of cells expressing +gene, or average expression scaled) will be returned, as selected by +user.
+Methodology
+This function creates a dot plot visualization of gene expression by
+metadata from a given dataset. It uses the Seurat package to create
+these plots. The size of the dot represents the percentage of cells
+expressing a particular gene (frequency), while the color of the dot
+indicates the average gene expression level. The function ensures that
+only unique and valid genes and categories are used. If some categories
+or genes are not found in the dataset, appropriate warnings are issued.
+The plot is then drawn with the option to reverse the x and y-axes and
+to reverse the order of metadata categories. The colors can also be
+customized. In addition to the plot, the function provides the tabular
+format of the dot plot data, which can be useful for further analysis or
+reporting. A choice of returning either the tables representing the
+percent of cells expressing a gene or the average expression level of
+the genes. This function can be useful for exploratory data analysis and
+visualizing the differences in gene expression across different
+conditions or groups of cells.
+
+FigOut=dotPlotMet(object=Anno_SO$object,
+ metadata="orig.ident",
+ cells=c("PBS","ENT","NHSIL12", "Combo","CD8dep" ),
+ markers=Marker_Table$Macrophages,
+ plot.reverse = FALSE,
+ cell.reverse.sort = FALSE,
+ dot.color = "darkblue"
+ )
-
+
Aran, D., A. P. Looney, L. Liu, E. Wu, V. Fong, A. Hsu, S. Chak, +et al. 2019. “Reference-based analysis of lung single-cell sequencing +reveals a transitional profibrotic macrophage.” Nat. Immunol. 20 (2): +163–72.
+http://bioconductor.org/packages/release/data/experiment/html/celldex.html
+- +
Hao Y et al. Integrated analysis of multimodal single-cell data. +Cell. 2021 Jun 24;184(13):3573-3587.e29. doi: +10.1016/j.cell.2021.04.048. Epub 2021 May 31. PMID: 34062119; PMCID: +PMC8238499.
+
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/SCWorkflow-QC.html b/docs/articles/SCWorkflow-QC.html
new file mode 100644
index 0000000..555c49e
--- /dev/null
+++ b/docs/articles/SCWorkflow-QC.html
@@ -0,0 +1,303 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Differential Expression Analysis
+ + + +SCWorkflow-DEG.Rmd
+
+
+
+
+
+DE with Find Markers +
++
This function performs a DE (differential expression) analysis on a +merged Seurat object to identify expression markers between different +groups of cells (contrasts). This analysis uses the FindMarkers() +function of the Seurat Workflow.
+Methodology Differential expression analysis (DEG) +is a fundamental technique in single-cell genomics research. The goal of +DEG analysis is to identify genes that exhibit significant changes in +expression levels between different groups of cells or conditions, +thereby uncovering potential markers that distinguish these groups. This +function takes a merged Seurat object [1] as input, expected to contain +single-cell data from multiple samples, along with relevant metadata and +SingleR annotations, which provide information about cell identity. To +perform the DEG analysis, the user can choose from various statistical +algorithms, such as MAST [2], wilcox [3], bimod [4], and more, which +accommodate different types of experimental designs and assumptions +about the data. The user can control the sensitivity of the analysis by +setting the minimum fold-change in gene expression between the groups to +be considered significant. Additionally, users can specify the assay to +be used for the analysis, whether it is the scaled data (SCT) or raw RNA +counts. For best results, it is recommended to use this function with +well-curated and preprocessed single-cell data, ensuring that the Seurat +object contains relevant metadata and SingleR annotations. Users should +carefully select the samples and contrasts based on their experimental +design and research questions. Additionally, exploring different +statistical algorithms and adjusting the threshold can fine-tune the DEG +analysis and reveal more accurate gene expression markers.
+
+
+DEG_table=degGeneExpressionMarkers(object = Anno_SO$object,
+ samples = c("PBS", "ENT", "NHSIL12", "Combo", "CD8dep" ),
+ contrasts = c("0-1"),
+ parameter.to.test = "SCT_snn_res_0_2",
+ test.to.use = "MAST",
+ log.fc.threshold = 0.25,
+ assay.to.use = "SCT",
+ use.spark = F
+ )| Gene | +C_0_vs_1_pval | +C_0_vs_1_logFC | +C_0_vs_1_pct1 | +C_0_vs_1_pct2 | +C_0_vs_1_adjpval | +
|---|---|---|---|---|---|
| Cd274 | +0 | +-1.1999485 | +0.369 | +0.808 | +0 | +
| Fth1 | +0 | +-1.4170103 | +1.000 | +1.000 | +0 | +
| Mmp12 | +0 | +-3.0512305 | +0.163 | +0.635 | +0 | +
| mt-Atp6 | +0 | +0.9790296 | +1.000 | +0.984 | +0 | +
| mt-Co2 | +0 | +1.0289905 | +1.000 | +0.982 | +0 | +
| mt-Co3 | +0 | +0.9775768 | +1.000 | +0.981 | +0 | +
-
+
- https://satijalab.org/seurat/ +
- https://rglab.github.io/MAST/ +
- Dalgaard, Peter (2008). Introductory Statistics with R. Springer +Science & Business Media. pp. 99–100 +
- https://en.wikipedia.org/wiki/Multimodal_distribution +
+
+
+ Pseudo Bulk Method +
++
+
+
+Aggregate Seurat Counts +
+This function is the first step in a Pseudobulk analysis of your +scRNA-seq dataset. It groups cells based on chosen categorical +variable(s) in the Seurat Object’s Metadata and aggregates the counts of +each gene in each group.
+The output is a table of aggregate expression in which the rows are +genes and the columns are values found in the chosen Pseudobulk +variable. If you select multiple categories to aggregate by +(e.g. Category1: A,B,C and Category2: D,E,F), cells will be grouped by +combinations of category variables (e.g. A_D, A_E, A_F, B_D, B_E, B_F). +By default, gene counts are averaged across cells in each group.
+
+AggOut=aggregateCounts(object=Anno_SO$object,
+ var.group=c( "orig.ident"),
+ slot="data")
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/SCWorkflow-SubsetReclust.html b/docs/articles/SCWorkflow-SubsetReclust.html
new file mode 100644
index 0000000..b901d0a
--- /dev/null
+++ b/docs/articles/SCWorkflow-SubsetReclust.html
@@ -0,0 +1,209 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Import Data and Quality Control
+ + + +SCWorkflow-QC.Rmd
+
+
+
+
+
+Process Input Data +
++
This package is designed to work with the general Seurat Workflow. To +begin using the SCWorkflow tools you will have to process the h5 files +generated by the Cell Ranger software from the 10x genomics platform to +create a list of Seurat Objects[1] corresponding to each h5 file. A +Seurat Object is the basic data structure for Seurat Single Cell +analysis
+This tool supports standard scRNAseq, CITE-Seq, and TCR-Seq assays. +Samples prepared with a cell hashing protocol (HTOs) can also be +processed to produce a Seurat Object split by the corresponding +experimental design strategy. h5 files containing multiple samples can +also be processed to create Seurat objects that will be split based on +the values in the orig.ident column.
+A corresponding Metadata table can be used to add sample level +information to the Seurat object. The table format should have Sample +names in the first Column and any sample metadata in additional columns. +The Metadata table can also be used to rename samples by including an +alternative sample name Column in the metadata table.
+| Sample_Name | +Rename | +Treatment | +
|---|---|---|
| SCAF1713_1_1 | +PBS | +WT | +
| SCAF1714_2_1 | +ENT | +Entinostat | +
| SCAF1715_3_1 | +NHSIL12 | +NHS-IL12 | +
| SCAF1716_4_1 | +Combo | +Entinostat + NHS-IL12 | +
| SCAF1717_5_1 | +CD8dep | +Entinostat + NHS-IL12 | +
Samples can also be excluded from the final Seurat object using a +REGEX strategy to identify the samples to be included/excluded. explain +based on newnames
+The final Seurat Object will contain an assay slot with log2 +normalized counts. QC figures for individual samples will also be +produced to help evaluate samples quality.
+
+
+SampleMetadataTable <- read.table(file = "./images/Sample_Metadata.txt", sep = '\t',header = T)
+files=list.files(path="../tests/testthat/fixtures/Chariou/h5files",full.names = T)
+
+SOlist=processRawData(input=files,
+ sample.metadata.table=SampleMetadataTable,
+ sample.name.column="Sample_Name",
+ organism="Mouse",
+ rename.col="Rename",
+ keep=T,
+ file.filter.regex=c(),
+ split.h5=F,
+ cell.hash=F,
+ tcr.summarize.topN=10,
+ do.normalize.data=T
+)

+
+
+
+Filter Low Quality Cells +
++
This function will filter genes and cells based on multiple metrics +available in the Seurat Object metadata slot. A detailed guide for +single cell quality filtering can be found from Xi and Li, 2021 [2]. +First, genes can be filtered by setting the minimum number of cells +needed to keep a gene or removing VDJ Add descriptiopn of VDJ genes. +Next, cells can be filtered by setting thresholds for each individual +metric. Cells that do not meet any of the designated criteria will be +removed from the final filtered Seurat Object . Filter limits can be set +by using absolute values or median absolute deviations (MADs) for each +criteria. If both absolute and MAD values are set for a single filter, +the least extreme value (i.e. the lowest value for upper limits or the +highest value for lower limits) will be selected. The filter values used +for each metric will be printed in the log output. All filters have +default values and can be turned off by setting limits to NA.
+The individual filtering criteria used in this tool are listed +below.
+-
+
- The total number of molecules detected within each cell +(nCount_RNA) +
- The number of genes detected in each cell (nFeature_RNA) +
- The complexity of of genes ( +log10(nFeature_RNA)/log10(nCount_RNA) +
- Percent of mitochondrial Genes +
- Percent counts in top 20 Genes +
- Doublets calculated by scDblFinder (using package default +parameters) [3] +
The function will return a filtered Seurat Object and various figures +showing metrics before and after filtering. These figures can be used to +help evaluate the effects of filtering criteria and whether filtering +limits need to be adjusted.
+
+
+SO_filtered=filterQC(object=SOlist$object,
+ ## Filter Genes
+ min.cells = 20,
+ filter.vdj.genes=FALSE,
+
+ ## Filter Cells
+ nfeature.limits=c(NA,NA),
+ mad.nfeature.limits=c(5,5),
+ ncounts.limits=c(NA,NA),
+ mad.ncounts.limits=c(5,5),
+ mitoch.limits = c(NA,25),
+ mad.mitoch.limits = c(NA,3),
+ complexity.limits = c(NA,NA),
+ mad.complexity.limits = c(5,NA),
+ topNgenes.limits = c(NA,NA),
+ mad.topNgenes.limits = c(5,5),
+ n.topgenes=20,
+ do.doublets.filter=TRUE
+ )


+
+
+
+ Combine, Normalize, and Cluster Data +
++
This functions combines multiple sample level Seurat Objects into a +single Seurat Object and normalizes the combined dataset. The +multi-dimensionality of the data will be summarized into a set of +“principal components” and visualized in both UMAP and tSNE projections. +A graph-based clustering approach will identify cell clusters with in +the data.
+
+
+Comb_SO=combineNormalize(
+ object=SO_filtered$object,
+ # Nomralization variables
+ npcs = 21,
+ SCT.level="Merged",
+ vars.to.regress = c("percent.mt"),
+ # FindVariableFeatures
+ nfeatures = 2000,
+ low.cut = 0.1,
+ high.cut = 8,
+ low.cut.disp = 1,
+ high.cut.disp = 100000,
+ selection.method = "vst",
+ # Dim Reduction
+ only.var.genes = FALSE,
+ draw.umap = TRUE,
+ draw.tsne = TRUE,
+ seed.for.pca = 42,
+ seed.for.tsne = 1,
+ seed.for.umap = 42,
+ # Clustering Varables
+ clust.res.low = 0.2,
+ clust.res.high = 1.2,
+ clust.res.bin = 0.2,
+ # Select PCs
+ methods.pca = NULL,
+ var.threshold = 0.1,
+ pca.reg.plot = FALSE,
+ jackstraw = FALSE,
+ jackstraw.dims=5,
+ # Other
+ exclude.sample = NULL,
+ cell.count.limit= 35000,
+ reduce.so = FALSE,
+ project.name = "scRNAProject",
+ cell.hashing.data = FALSE
+)

-
+
Hao Y et al. Integrated analysis of multimodal single-cell data. +Cell. 2021 Jun 24;184(13):3573-3587.e29. doi: +10.1016/j.cell.2021.04.048. Epub 2021 May 31. PMID: 34062119; PMCID: +PMC8238499.
+Heumos, L., Schaar, A.C., Lance, C. et al. Best practices for +single-cell analysis across modalities. Nat Rev Genet (2023). https://doi.org/10.1038/s41576-023-00586-w
+Germain P, Lun A, Macnair W, Robinson M (2021). “Doublet +identification in single-cell sequencing data using scDblFinder.” +f1000research. doi:10.12688/f1000research.73600.1.
+
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/SCWorkflow-Usage.html b/docs/articles/SCWorkflow-Usage.html
new file mode 100644
index 0000000..f46448f
--- /dev/null
+++ b/docs/articles/SCWorkflow-Usage.html
@@ -0,0 +1,200 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Subset and Recluster
+ + + +SCWorkflow-SubsetReclust.Rmd
+
+
+
+Subset Seurat Object +
++
This function subsets your Seurat object. Select a metadata column +and values matching the cells to pass forward in your analysis.
+
+
+filter_SO=filterSeuratObjectByMetadata(
+ object = Anno_SO$object,
+ samples.to.include = c("PBS","ENT","NHSIL12","Combo","CD8dep"),
+ sample.name = "orig.ident",
+ category.to.filter = "immgen_main",
+ values.to.filter = c("Monocytes","Macrophages","DC"),
+ keep.or.remove = FALSE,
+ greater.less.than = "greater than",
+ colors = c(
+ "aquamarine3",
+ "salmon1",
+ "lightskyblue3"),
+ seed = 10,
+ cut.off = 0.5,
+ legend.position = "right",
+ reduction = "umap",
+ plot.as.interactive.plot = FALSE,
+ legend.symbol.size = 2,
+ dot.size = 0.1,
+ number.of.legend.columns = 1,
+ dot.size.highlighted.cells = 0.5,
+ use.cite.seq.data = FALSE
+ ) 
+
+
+
+ Recluster Seurat Object +
++
This function provides a mechanism for re-clustering a filtered +Seurat object, a common task in single-cell RNA sequencing analysis. The +function provides options to choose the number of principal components, +range of clustering resolution, type of dimensionality reduction, and +several other parameters. The function finds variable features and +performs Principal Component Analysis (PCA). Next, dimensionality +reduction is performed using UMAP and t-SNE based on PCA, followed by +identification of nearest neighbors. It then performs clustering at +different resolutions within the provided range, creating new clustering +columns in the Seurat object. It also retains the old clustering +information, plots the clusters for each resolution and returns a list +containing the re-clustered Seurat object and the grid of clustering +plots. This function can be helpful for experimenting with different +clustering parameters especially after filtering and visually inspect +the results.
+Methodology
+This function uses methods from the Seurat package [1].
+Seurat uses a graph-based clustering method inspired by previous
+strategies, particularly those of Macosko et al [3]. It uses methods
+like SNN-Cliq and PhenoGraph [4.5], which represent cells in a graph
+structure based on similarities in feature expression patterns. The aim
+is to divide this graph into highly connected communities or clusters.
+The process begins by building a K-nearest neighbor (KNN) graph using
+Euclidean distance in PCA space. The algorithm refines the edge weights
+between cells according to their local neighborhood overlap, calculated
+using the Jaccard similarity measure. This is performed using predefined
+dimensions of the dataset, such as the first 10 Principal Components
+(PCs). To cluster cells, Seurat uses modularity optimization techniques
+like the Louvain [4] algorithm or SLM [5]. The ‘resolution’ parameter
+can be adjusted to control the granularity of the downstream clustering;
+a higher resolution results in more clusters. For single-cell datasets
+of approximately 3K cells, the recommended range for this parameter is
+between 0.4 and 1.2, but larger datasets typically require a higher
+resolution.
+
+reClust_SO=reclusterSeuratObject(
+ object = filter_SO$object,
+ prepend.txt = "old",
+ old.columns.to.save=c("orig_ident","Sample_Name","nCount_RNA","nFeature_RNA","percent_mt",
+ "log10GenesPerUMI","S_Score","G2M_Score","Phase","CC_Difference","Treatment",
+ "pct_counts_in_top_N_genes","Doublet","nCount_SCT","nFeature_SCT",
+ "mouseRNAseq_main","mouseRNAseq","immgen_main","immgen" ),
+ number.of.pcs = 50,
+ cluster.resolution.low.range = 0.2,
+ cluster.resolution.high.range = 1.2,
+ cluster.resolution.range.bins = 0.2,
+ reduction.type = "umap"
+ )
-
+
Seurat Clustering method https://satijalab.org/seurat/articles/pbmc3k_tutorial.html
+Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, +Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, +Sanes JR, Shalek AK, Regev A, McCarroll SA. Highly Parallel Genome-wide +Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. +2015 May 21;161(5):1202-1214.
+Xu, Chen, and Zhengchang Su. Identification of cell types from +single-cell transcriptomes using a novel clustering method. +Bioinformatics 31.12 (2015): 1974-1980.
+Levine, Jacob H., et al. Data-driven phenotypic dissection of AML +reveals progenitor-like cells that correlate with prognosis. Cell 162.1 +(2015): 184-197.
+Blondel, Vincent D., et al. Fast unfolding of communities in +large networks.”Journal of statistical mechanics: theory and experiment +2008.10 (2008): P10008.
+Waltman, Ludo, and Nees Jan Van Eck. A smart local moving +algorithm for large-scale modularity-based community detection. The +European physical journal B 86 (2013): 1-14
+
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/SCWorkflow-Visualizations.html b/docs/articles/SCWorkflow-Visualizations.html
new file mode 100644
index 0000000..deb01e0
--- /dev/null
+++ b/docs/articles/SCWorkflow-Visualizations.html
@@ -0,0 +1,515 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Getting Started
+ + + +SCWorkflow-Usage.Rmd
+
+
+Use SCWorkflow Container +
++
The SCWorkflow docker container is available on Biowulf which can be +used with RStudio to organize and rune the SCWorkflow package.
+You will need 2 shells (terminals) to set up RStudio on Biowulf.
+
+
+
+1. Log in to Biowulf +
+Open a terminal and login to biowulf then call an interactive +session
+ssh username@helix.nih.gov
+
+sinteractive --tunnel --time=12:00:00 --mem=50g --cpus-per-task=16 --gres=lscratch:50
+
+
+
+
+3. Load the Container +
+The single cell container will emulate the environments from +NIDAP
+
+source /data/CCBR/NIDAP/container_singlecell.sh
+
+ + + + +
+Copy Files from Rstuido server to Helix +
++ + + + +
+
+
+ Install Package +
++
for general use SCWorkflow can be installed into your Rlibrary
+# install.packages("remotes")
+# remotes::install_github("NIDAP-Community/SCWorkflow", dependencies = TRUE)
+
+library(SCWorkflow)
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/articles/images/Anno1.png b/docs/articles/images/Anno1.png
new file mode 100644
index 0000000..77aadc5
Binary files /dev/null and b/docs/articles/images/Anno1.png differ
diff --git a/docs/articles/images/Anno2.png b/docs/articles/images/Anno2.png
new file mode 100644
index 0000000..07b6c6e
Binary files /dev/null and b/docs/articles/images/Anno2.png differ
diff --git a/docs/articles/images/CBM1.png b/docs/articles/images/CBM1.png
new file mode 100644
index 0000000..068a006
Binary files /dev/null and b/docs/articles/images/CBM1.png differ
diff --git a/docs/articles/images/CN1.png b/docs/articles/images/CN1.png
new file mode 100644
index 0000000..3943371
Binary files /dev/null and b/docs/articles/images/CN1.png differ
diff --git a/docs/articles/images/CN2.png b/docs/articles/images/CN2.png
new file mode 100644
index 0000000..54e40f8
Binary files /dev/null and b/docs/articles/images/CN2.png differ
diff --git a/docs/articles/images/DEV_CheatSheet.png b/docs/articles/images/DEV_CheatSheet.png
new file mode 100644
index 0000000..ea888ee
Binary files /dev/null and b/docs/articles/images/DEV_CheatSheet.png differ
diff --git a/docs/articles/images/DL1.png b/docs/articles/images/DL1.png
new file mode 100644
index 0000000..6c7a940
Binary files /dev/null and b/docs/articles/images/DL1.png differ
diff --git a/docs/articles/images/DL2.png b/docs/articles/images/DL2.png
new file mode 100644
index 0000000..d115a0c
Binary files /dev/null and b/docs/articles/images/DL2.png differ
diff --git a/docs/articles/images/DPM.png b/docs/articles/images/DPM.png
new file mode 100644
index 0000000..b046d7a
Binary files /dev/null and b/docs/articles/images/DPM.png differ
diff --git a/docs/articles/images/MS1.png b/docs/articles/images/MS1.png
new file mode 100644
index 0000000..d601811
Binary files /dev/null and b/docs/articles/images/MS1.png differ
diff --git a/docs/articles/images/MS2.png b/docs/articles/images/MS2.png
new file mode 100644
index 0000000..46e88d4
Binary files /dev/null and b/docs/articles/images/MS2.png differ
diff --git a/docs/articles/images/MS3.png b/docs/articles/images/MS3.png
new file mode 100644
index 0000000..b447d4d
Binary files /dev/null and b/docs/articles/images/MS3.png differ
diff --git a/docs/articles/images/ProcessInputData1.png b/docs/articles/images/ProcessInputData1.png
new file mode 100644
index 0000000..96c1dbe
Binary files /dev/null and b/docs/articles/images/ProcessInputData1.png differ
diff --git a/docs/articles/images/ProcessInputData2.png b/docs/articles/images/ProcessInputData2.png
new file mode 100644
index 0000000..12ecdb3
Binary files /dev/null and b/docs/articles/images/ProcessInputData2.png differ
diff --git a/docs/articles/images/QC1.png b/docs/articles/images/QC1.png
new file mode 100644
index 0000000..3b30e3f
Binary files /dev/null and b/docs/articles/images/QC1.png differ
diff --git a/docs/articles/images/QC2.png b/docs/articles/images/QC2.png
new file mode 100644
index 0000000..951361a
Binary files /dev/null and b/docs/articles/images/QC2.png differ
diff --git a/docs/articles/images/QC3.png b/docs/articles/images/QC3.png
new file mode 100644
index 0000000..567ff97
Binary files /dev/null and b/docs/articles/images/QC3.png differ
diff --git a/docs/articles/images/RNC.png b/docs/articles/images/RNC.png
new file mode 100644
index 0000000..c849208
Binary files /dev/null and b/docs/articles/images/RNC.png differ
diff --git a/docs/articles/images/SubRec_recl.png b/docs/articles/images/SubRec_recl.png
new file mode 100644
index 0000000..81ad2eb
Binary files /dev/null and b/docs/articles/images/SubRec_recl.png differ
diff --git a/docs/articles/images/SubRec_sub2.png b/docs/articles/images/SubRec_sub2.png
new file mode 100644
index 0000000..6a1db85
Binary files /dev/null and b/docs/articles/images/SubRec_sub2.png differ
diff --git a/docs/articles/images/Vis_3D.html b/docs/articles/images/Vis_3D.html
new file mode 100644
index 0000000..182a5b0
--- /dev/null
+++ b/docs/articles/images/Vis_3D.html
@@ -0,0 +1,1945 @@
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Visualizations
+ + + +SCWorkflow-Visualizations.Rmd
+
+
+
+Color by Metadata +
++
Use this Function to color your dimensionality reduction (TSNE & +UMAP) with different columns from your Metadata Table. You can select +one or more columns from the Metadata Table, and for each column +selected, this function will produce a plot (t-SNE & UMAP) using the +data in that column to color the cells.
+This function visualizes your plot based on selected metadata. Users +can customize how they want to visualize the data, including the type of +visualization used, the size and shape of the points, and the level of +transparency.
+
+FigOut=plotMetadata(
+ object=Anno_SO$object,
+ samples.to.include=c("PBS","ENT","NHSIL12","Combo","CD8dep" ),
+ metadata.to.plot=c("SCT_snn_res.0.4","Phase"),
+ columns.to.summarize=NULL,
+ summarization.cut.off = 5,
+ reduction.type = "umap",
+ use.cite.seq = FALSE,
+ show.labels = FALSE,
+ legend.text.size = 1,
+ legend.position = "right",
+ dot.size = 0.01
+ )
+
+
+
+Plot 3D Dimensionality Reduction +
++
This Function creates 3D interactive UMAP or t-SNE plot. The plot is +saved in the output folder as an HTML file that can be downloaded.
+This function is designed to generate a 3D t-SNE visualization based +on a given Seurat Object. The output includes an interactive plot and a +dataframe containing the t-SNE coordinates. The function accepts several +parameters, such as the Seurat Object, a metadata column for color, a +metadata column for labeling, dot size for the plot, legend display +option, colors for the color variable, filename for saving the plot, the +number of principal components for t-SNE calculations, and an option to +save the plot as a widget in an HTML file. Initially, the function +executes t-SNE on the Seurat Object to obtain the 3D coordinates. +Subsequently, it constructs a dataframe for the Plotly visualization, +incorporating the t-SNE coordinates, color variable, and label variable. +The function generates a 3D scatter plot using the t-SNE coordinates. +Finally, the function saves the plot as an embedded Plotly image in an +HTML file.
+Methodology
+t-Distributed Stochastic Neighbor Embedding (t-SNE) is a sophisticated
+dimensionality reduction technique frequently employed for the
+visualization of high-dimensional data [1]. It effectively displays the
+relationships between individual cells based on their gene expression
+profiles. To compute t-SNE, the algorithm constructs a probability
+distribution representing similarities between data points in
+high-dimensional space. Subsequently, it generates a lower-dimensional
+representation, typically two or three dimensions, wherein the distances
+between data points reflect their similarities in the high-dimensional
+space. The algorithm employs an iterative process to adjust the
+positions of cells in a lower-dimensional space, aiming to minimize
+discrepancies between the original high-dimensional similarities and
+those in the lower-dimensional space. This approach enables the
+algorithm to capture both global and local structures within the data,
+effectively revealing clusters or groups of similar cells.
+
+FigOut=tSNE3D(
+ object=Anno_SO$object,
+ color.variable="SCT_snn_res.0.4",
+ label.variable="SCT_snn_res.0.4",
+ dot.size = 4,
+ legend = TRUE,
+ colors = c("darkblue","purple4","green","red","darkcyan",
+ "magenta2","orange","yellow","black"),
+ filename = "plot.html",
+ save.plot = FALSE,
+ npcs = 15
+ )
+
+
+
+Color by Genes +
++
This function visualizes gene expression intensities for provided +Genes across your cells. If a gene is not found in the dataset, the Log +will report this. Otherwise, you should see one plot (TSNE or UMAP, your +choice) per gene name provided. The intensity of the red color will be +relative to the expression of the gene in that cell. Final Potomac +Compatible Version: v98. Sugarloaf V1: v103. [View
+Methodology
+This function visualizes expression values of the chosen gene or protein
+in different samples. Users can customize how they want to visualize the
+data, including the type of visualization used, the size and shape of
+the points, and the level of transparency.
+
+FigOut=colorByGene(
+ object=Anno_SO$object,
+ samples.to.include=c("PBS","ENT","NHSIL12","Combo","CD8dep" ),
+ gene="Itgam",
+ reduction.type = "umap",
+ number.of.rows = 0,
+ return.seurat.object = FALSE,
+ color = "red",
+ point.size = 1,
+ point.shape = 16,
+ point.transparency = 0.5,
+ use.cite.seq.data = FALSE,
+ assay = "SCT")
+
+
+
+Violin Plot from Seurat Object +
++
This Function allows for the generation of customized violin plots to +visualize transcriptional changes and interactions in single-cell +RNA-seq data, providing insights into the cellular heterogeneity and +dynamics within the dataset.
+Methodology
+The Function organizes your data based on specific groups that you
+choose from a Seurat Object metadata. It then gathers information about
+the activity levels of specific genes you’re interested in. You can, if
+you wish, change the names or the order of these groups based on a
+column you specify in the data. This feature lets you tailor the
+analysis more closely to your needs. The code also has a function that
+removes any odd data points that might distort the results, and it
+adjusts the data to make it easier to visualize through jittered points
+and an overlaying boxplot displaying quantile information. Then, the
+code creates violin plots, which allows you to see how the activity
+levels of genes vary within each group [2]. The graph is customizable,
+letting you set various options such as limit values on the vertical
+axis, displaying individual data points, converting scales to
+logarithmic, and showing boxplots. You can choose how the plot looks -
+whether it’s laid out like a grid, in rows, or with customized
+labels.
+
+FigOut=violinPlot(
+ object=Anno_SO$object,
+ assay="SCT",
+ layer="scale.data",
+ genes=c("Itgam","Cd38"),
+ group="SCT_snn_res.0.4",
+ facet.by = "",
+ filter.outliers = F,
+ outlier.low = 0.05,
+ outlier.high = 0.95,
+ jitter.points = TRUE,
+ jitter.dot.size = 1
+ )
+
+
+
+Heatmap +
++
This Function provides a comprehensive method for visualizing single
+cell transcript and/or protein expression data in the form of a heatmap.
+The data is obtained from a Seurat object, and the user can specify a
+set of genes for the analysis.
+The Function allows optional ordering by metadata (categorical) or
+gene/protein expression levels. Visualization customization options
+include color choices for the heatmap, addition of gene or protein
+annotations, and optional arrangement by metadata.
Key features include: - Options for adding gene or protein +annotations, metadata arrangement, and specifying row and column names. +- Customizable visualization settings including font sizes for rows, +columns, and legend, row height, and heatmap colors. - Ability to trim +outlier data, perform z-scaling on rows, and set row order.
+The Function returns a heatmap plot along with the underlying data +used to generate it. It also allows the user to set a seed for color +generation and specify outlier data parameters. This function is +particularly useful for exploratory data analysis and preliminary data +visualization in single cell studies.
+Methodology
+In this method, two hierarchical clustering processes are performed: one
+on the rows and one on the columns of the dataset unless ordered by
+annotations. Hierarchical clustering is a method of cluster analysis
+that aims to build a hierarchy of clusters. The result is a tree-like
+diagram called a dendrogram, where similar data points (e.g., genes or
+samples) are joined together into clusters at the “branches”, based on a
+mathematical measure of similarity such as Euclidean or Manhattan
+distance. The heatmap is produced by a package called ComplexHeatmap [3]
+and presents the data matrix where rows represent individual genes (or
+proteins, metabolites, etc.) and columns represent different samples
+(e.g., tissue samples, cells, experimental conditions). The color at
+each position in the grid corresponds to the expression level of a gene
+in a particular sample, with one color representing upregulation (higher
+expression), another representing downregulation (lower expression), and
+usually a neutral color representing no change. This allows for easy
+visual interpretation of patterns or correlations in the data.
+
+FigOut=heatmapSC(
+ object=Anno_SO$object,
+ sample.names=c("PBS","ENT","NHSIL12","Combo","CD8dep" ),
+ metadata="SCT_snn_res.0.4",
+ transcripts=c("Cd163","Cd38","Itgam","Cd4","Cd8a","Pdcd1","Ctla4"),
+ use.assay = "SCT",
+ proteins = NULL,
+ heatmap.color = "Bu Yl Rd",
+ plot.title = "Heatmap",
+ add.gene.or.protein = FALSE,
+ protein.annotations = NULL,
+ rna.annotations = NULL,
+ arrange.by.metadata = TRUE,
+ add.row.names = TRUE,
+ add.column.names = FALSE,
+ row.font = 5,
+ col.font = 5,
+ legend.font = 5,
+ row.height = 15,
+ set.seed = 6,
+ scale.data = TRUE,
+ trim.outliers = TRUE,
+ trim.outliers.percentage = 0.01,
+ order.heatmap.rows = FALSE,
+ row.order = c()
+ )
+
+
+
+Dot Plot of Genes by Metadata +
++
This function creates a dot plot of average gene expression values +for a set of genes in cell subpopulations defined by metadata annotation +columns. The input table contains a single column for genes (the “Genes +column”) and a single column for category (the “Category labels to plot” +column). The values in the “Category labels to plot” column should match +the values provided in the metadata function (Metadata Category to +Plot). The plot will order the genes (x-axis, left to right) and +Categories (y-axis, top to bottom) in the order in which it appears in +the input table. Any category entries omitted will not be plotted.
+The Dotplot size will reflect the percentage of cells expressing the +gene while the color will reflect the average expression for the gene. A +table showing values on the plot (either percentage of cells expressing +gene, or average expression scaled) will be returned, as selected by +user.
+Methodology
+This function creates a dot plot visualization of gene expression by
+metadata from a given dataset. It uses the Seurat package to create
+these plots. The size of the dot represents the percentage of cells
+expressing a particular gene (frequency), while the color of the dot
+indicates the average gene expression level. The function ensures that
+only unique and valid genes and categories are used. If some categories
+or genes are not found in the dataset, appropriate warnings are issued.
+The plot is then drawn with the option to reverse the x and y-axes and
+to reverse the order of metadata categories. The colors can also be
+customized. In addition to the plot, the function provides the tabular
+format of the dot plot data, which can be useful for further analysis or
+reporting. A choice of returning either the tables representing the
+percent of cells expressing a gene or the average expression level of
+the genes. This function can be useful for exploratory data analysis and
+visualizing the differences in gene expression across different
+conditions or groups of cells.
-
+
- Seurat package Dotplot Documentation https://satijalab.org/seurat/reference/dotplot + +
+
+FigOut=dotPlotMet(
+ object=Anno_SO$object,
+ metadata="SCT_snn_res.0.4",
+ cells=unique(Anno_SO$object$SCT_snn_res.0.4),
+ markers=c("Itgam","Cd163","Cd38","Cd4","Cd8a","Pdcd1","Ctla4"),
+ plot.reverse = FALSE,
+ cell.reverse.sort = FALSE,
+ dot.color = "darkblue"
+ )
+
+
+
+ Compare Cell Populations +
++
This function compares cell population composition across +experimental groups (for example sample, treatment, timepoints, or donor +cohorts) using metadata already stored in the Seurat object. It is +useful after clustering and annotation, when you want to quantify how +specific cell populations shift between conditions. + The function supports both Frequency (percent) and +Counts (absolute cell numbers) modes. In most +biological comparisons with unequal total cell recovery across samples, +frequency mode is preferred for interpretation. Counts mode can be +useful for QC and yield-focused assessments.
+ +Methodology
+The method first aggregates metadata by annotation and group to compute
+percentages and counts. It then links these summaries to sample-level
+metadata and generates a composition-focused barplot for sample-level
+variability. Together, these plots help distinguish overall
+compositional shifts from replicate-level dispersion.
+
+FigOut=compareCellPopulations(
+ object=Anno_SO$object,
+ annotation.column="immgen_main",
+ group.column="Treatment",
+ sample.column = "orig.ident",
+ counts.type = "Frequency",
+ group.order = NULL,
+ wrap.ncols = 5
+)
+ 
-
+
Seurat Documentation for t-SNE Analysis https://satijalab.org/seurat/reference/runtsne
+- +
Complex Heatmap Reference Book https://jokergoo.github.io/ComplexHeatmap-reference/book/
+
+
+
+
+
+
+
+ + + + +
+
+
+
+
+
+
diff --git a/docs/articles/images/Vis_CBG.png b/docs/articles/images/Vis_CBG.png
new file mode 100644
index 0000000..453044b
Binary files /dev/null and b/docs/articles/images/Vis_CBG.png differ
diff --git a/docs/articles/images/Vis_CBM.png b/docs/articles/images/Vis_CBM.png
new file mode 100644
index 0000000..a0bf1ca
Binary files /dev/null and b/docs/articles/images/Vis_CBM.png differ
diff --git a/docs/articles/images/Vis_CCPbar.png b/docs/articles/images/Vis_CCPbar.png
new file mode 100644
index 0000000..46f47d3
Binary files /dev/null and b/docs/articles/images/Vis_CCPbar.png differ
diff --git a/docs/articles/images/Vis_CCPbox.png b/docs/articles/images/Vis_CCPbox.png
new file mode 100644
index 0000000..94c402c
Binary files /dev/null and b/docs/articles/images/Vis_CCPbox.png differ
diff --git a/docs/articles/images/Vis_DEGAggBar.png b/docs/articles/images/Vis_DEGAggBar.png
new file mode 100644
index 0000000..9c77538
Binary files /dev/null and b/docs/articles/images/Vis_DEGAggBar.png differ
diff --git a/docs/articles/images/Vis_DPM.png b/docs/articles/images/Vis_DPM.png
new file mode 100644
index 0000000..a994770
Binary files /dev/null and b/docs/articles/images/Vis_DPM.png differ
diff --git a/docs/articles/images/Vis_HM.png b/docs/articles/images/Vis_HM.png
new file mode 100644
index 0000000..cb4f360
Binary files /dev/null and b/docs/articles/images/Vis_HM.png differ
diff --git a/docs/articles/images/Vis_Violin.png b/docs/articles/images/Vis_Violin.png
new file mode 100644
index 0000000..43e0338
Binary files /dev/null and b/docs/articles/images/Vis_Violin.png differ
diff --git a/docs/articles/index.html b/docs/articles/index.html
new file mode 100644
index 0000000..25cafbf
--- /dev/null
+++ b/docs/articles/index.html
@@ -0,0 +1,89 @@
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/authors.html b/docs/authors.html
new file mode 100644
index 0000000..538b72b
--- /dev/null
+++ b/docs/authors.html
@@ -0,0 +1,112 @@
+
+
+
+
+
+
+
+
+
+ Articles
+
+
+ Vignettes
+ + +
+
+
+
+ Developer
+Developer documentation
+
+
+
+
+
+
+
+
diff --git a/docs/decision_log.html b/docs/decision_log.html
new file mode 100644
index 0000000..8803193
--- /dev/null
+++ b/docs/decision_log.html
@@ -0,0 +1,78 @@
+
+
+
+
+
+
+
+
+
+SCWorkflow AI Coding Instructions
+ +
+
+
+
+
+
+Project Overview
+SCWorkflow is an R package for single-cell RNA-seq analysis built on the Seurat framework. It’s designed for analyzing multimodal 10x Genomics data, with support for CITE-Seq, cell hashing, and TCR-seq data. The package is deployed as both an R package and Docker container for use in NIDAP (Palantir Foundry) and Biowulf HPC environments.
+
+
+Core Architecture
+
+
+Sequential Workflow Pattern
+Functions follow a numbered workflow sequence: 1. processRawData() - Process H5 files into Seurat objects 2. filterQC() - Quality control and filtering
+3. combineNormalize() - Merge samples, normalize, dimension reduction 4. Harmony integration (optional) - Batch correction 5. annotateCellTypes() - Automatic cell type annotation via SingleR 6. Analysis functions - DEG, visualization, clustering
+
+Seurat Object as Central Data Structure
+- All functions expect/return Seurat objects as primary data containers +
- Functions often modify objects in-place and return modified versions +
- Metadata is heavily used for sample tracking and analysis parameters +
- Multiple assays supported: RNA, SCT (SCTransform), ADT (CITE-seq), etc. +
+
+Function Design Patterns
+- +Comprehensive parameter lists: Most functions have 15-30+ parameters with sensible defaults +
- +Conditional workflows: Functions check input types (H5 vs Seurat) and adapt behavior +
- +Multi-sample handling: Functions can process lists of Seurat objects or file paths +
- +Plotting integration: Most analysis functions return both data and visualization outputs +
+
+
+Key Conventions
+
+
+File Organization
+-
+
R/contains one function per file, named descriptively (e.g.,Process_Raw_Data.R)
+ - Function names use camelCase:
processRawData(),annotateCellTypes()+
+ - File names use Snake_Case with capitalization:
Process_Raw_Data.R+
+
+
+
+Parameter Naming Patterns
+-
+
object- Primary Seurat object input
+ -
+
samples.to.include- Character vector for sample subsetting
+ -
+
reduction.type- Visualization method (“umap”, “tsne”, “pca”)
+ -
+
organism- Species specification (“Human” or “Mouse”)
+ - Boolean parameters use
.separator:do.normalize.data,draw.umap+
+
+
+Output standards
+- Generate R CMD check friendly code. +
- Use roxygen2 for exported functions. +
- Add utils::globalVariables() for NSE variables. +
- Prefer explicit namespaces (dplyr::, ggplot2::). +
- Avoid side effects in tests. +
+
+Template conventions
+- JSON templates live under inst/extdata/NIDAPjson/. +
- JSON is the source-of-truth for arguments, defaults, and behavior. +
- orderedMustacheKeys defines argument order. +
+
+Testing Structure
+- Comprehensive
tests/testthat/with both unit tests and integration tests
+ -
+
fixtures/directory contains real Seurat objects for testing
+ - Helper functions in
helper-*.Rfiles for test setup
+ - Tests follow naming convention:
test-Function_Name.R+
+
+
+
+
+Deliverables
+- Update CHANGELOG.md, decision_log.md, docs/session_notes.md. +
- Add testthat tests for each generated function. +
+
+Critical Dependencies and Integration Points
+
+
+
+
+Bioconductor/Seurat Ecosystem
+- +Seurat 4.1.1+: Core framework for single-cell analysis +
- +SingleR: Automated cell type annotation +
- +MAST/limma/edgeR: Differential expression analysis +
- +Harmony: Batch effect correction and integration +
+
+Common Debugging Areas
+
+
+
+
+Data Type Handling
+- Functions check for H5 vs Seurat object inputs:
is(input, "Seurat")+
+ - Cell filtering can drastically reduce object sizes - verify cell counts +
- Assay availability: functions may require specific assays (RNA, SCT, ADT) +
+
+Quick Start for AI Agents
+When working with this codebase: 1. Always check if input is a Seurat object: class(object) 2. Verify required reductions exist: object@reductions 3. Check available assays: names(object@assays) 4. Use str(object@meta.data) to understand metadata structure 5. Test functions start with small datasets from tests/testthat/fixtures/
+
+
+
+
+
+
+
+
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmQiArmlw.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmQiArmlw.woff2
new file mode 100644
index 0000000..b0ed6d6
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmQiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmUiAo.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmUiAo.woff2
new file mode 100644
index 0000000..5a5fad1
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmUiAo.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmXiArmlw.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmXiArmlw.woff2
new file mode 100644
index 0000000..de646f8
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmXiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmYiArmlw.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmYiArmlw.woff2
new file mode 100644
index 0000000..6a6fb7a
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmYiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmZiArmlw.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmZiArmlw.woff2
new file mode 100644
index 0000000..1a1ed7c
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmZiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmaiArmlw.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmaiArmlw.woff2
new file mode 100644
index 0000000..47e69cf
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmaiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmbiArmlw.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmbiArmlw.woff2
new file mode 100644
index 0000000..bc95855
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmbiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVn6iArmlw.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVn6iArmlw.woff2
new file mode 100644
index 0000000..f9c26fa
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVn6iArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVnoiArmlw.woff2 b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVnoiArmlw.woff2
new file mode 100644
index 0000000..15e1583
Binary files /dev/null and b/docs/deps/Roboto-0.4.10/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVnoiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.10/font.css b/docs/deps/Roboto-0.4.10/font.css
new file mode 100644
index 0000000..33cb5f4
--- /dev/null
+++ b/docs/deps/Roboto-0.4.10/font.css
@@ -0,0 +1,90 @@
+/* cyrillic-ext */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmZiArmlw.woff2) format('woff2');
+ unicode-range: U+0460-052F, U+1C80-1C8A, U+20B4, U+2DE0-2DFF, U+A640-A69F, U+FE2E-FE2F;
+}
+/* cyrillic */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmQiArmlw.woff2) format('woff2');
+ unicode-range: U+0301, U+0400-045F, U+0490-0491, U+04B0-04B1, U+2116;
+}
+/* greek-ext */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmYiArmlw.woff2) format('woff2');
+ unicode-range: U+1F00-1FFF;
+}
+/* greek */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmXiArmlw.woff2) format('woff2');
+ unicode-range: U+0370-0377, U+037A-037F, U+0384-038A, U+038C, U+038E-03A1, U+03A3-03FF;
+}
+/* math */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVnoiArmlw.woff2) format('woff2');
+ unicode-range: U+0302-0303, U+0305, U+0307-0308, U+0310, U+0312, U+0315, U+031A, U+0326-0327, U+032C, U+032F-0330, U+0332-0333, U+0338, U+033A, U+0346, U+034D, U+0391-03A1, U+03A3-03A9, U+03B1-03C9, U+03D1, U+03D5-03D6, U+03F0-03F1, U+03F4-03F5, U+2016-2017, U+2034-2038, U+203C, U+2040, U+2043, U+2047, U+2050, U+2057, U+205F, U+2070-2071, U+2074-208E, U+2090-209C, U+20D0-20DC, U+20E1, U+20E5-20EF, U+2100-2112, U+2114-2115, U+2117-2121, U+2123-214F, U+2190, U+2192, U+2194-21AE, U+21B0-21E5, U+21F1-21F2, U+21F4-2211, U+2213-2214, U+2216-22FF, U+2308-230B, U+2310, U+2319, U+231C-2321, U+2336-237A, U+237C, U+2395, U+239B-23B7, U+23D0, U+23DC-23E1, U+2474-2475, U+25AF, U+25B3, U+25B7, U+25BD, U+25C1, U+25CA, U+25CC, U+25FB, U+266D-266F, U+27C0-27FF, U+2900-2AFF, U+2B0E-2B11, U+2B30-2B4C, U+2BFE, U+3030, U+FF5B, U+FF5D, U+1D400-1D7FF, U+1EE00-1EEFF;
+}
+/* symbols */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVn6iArmlw.woff2) format('woff2');
+ unicode-range: U+0001-000C, U+000E-001F, U+007F-009F, U+20DD-20E0, U+20E2-20E4, U+2150-218F, U+2190, U+2192, U+2194-2199, U+21AF, U+21E6-21F0, U+21F3, U+2218-2219, U+2299, U+22C4-22C6, U+2300-243F, U+2440-244A, U+2460-24FF, U+25A0-27BF, U+2800-28FF, U+2921-2922, U+2981, U+29BF, U+29EB, U+2B00-2BFF, U+4DC0-4DFF, U+FFF9-FFFB, U+10140-1018E, U+10190-1019C, U+101A0, U+101D0-101FD, U+102E0-102FB, U+10E60-10E7E, U+1D2C0-1D2D3, U+1D2E0-1D37F, U+1F000-1F0FF, U+1F100-1F1AD, U+1F1E6-1F1FF, U+1F30D-1F30F, U+1F315, U+1F31C, U+1F31E, U+1F320-1F32C, U+1F336, U+1F378, U+1F37D, U+1F382, U+1F393-1F39F, U+1F3A7-1F3A8, U+1F3AC-1F3AF, U+1F3C2, U+1F3C4-1F3C6, U+1F3CA-1F3CE, U+1F3D4-1F3E0, U+1F3ED, U+1F3F1-1F3F3, U+1F3F5-1F3F7, U+1F408, U+1F415, U+1F41F, U+1F426, U+1F43F, U+1F441-1F442, U+1F444, U+1F446-1F449, U+1F44C-1F44E, U+1F453, U+1F46A, U+1F47D, U+1F4A3, U+1F4B0, U+1F4B3, U+1F4B9, U+1F4BB, U+1F4BF, U+1F4C8-1F4CB, U+1F4D6, U+1F4DA, U+1F4DF, U+1F4E3-1F4E6, U+1F4EA-1F4ED, U+1F4F7, U+1F4F9-1F4FB, U+1F4FD-1F4FE, U+1F503, U+1F507-1F50B, U+1F50D, U+1F512-1F513, U+1F53E-1F54A, U+1F54F-1F5FA, U+1F610, U+1F650-1F67F, U+1F687, U+1F68D, U+1F691, U+1F694, U+1F698, U+1F6AD, U+1F6B2, U+1F6B9-1F6BA, U+1F6BC, U+1F6C6-1F6CF, U+1F6D3-1F6D7, U+1F6E0-1F6EA, U+1F6F0-1F6F3, U+1F6F7-1F6FC, U+1F700-1F7FF, U+1F800-1F80B, U+1F810-1F847, U+1F850-1F859, U+1F860-1F887, U+1F890-1F8AD, U+1F8B0-1F8BB, U+1F8C0-1F8C1, U+1F900-1F90B, U+1F93B, U+1F946, U+1F984, U+1F996, U+1F9E9, U+1FA00-1FA6F, U+1FA70-1FA7C, U+1FA80-1FA89, U+1FA8F-1FAC6, U+1FACE-1FADC, U+1FADF-1FAE9, U+1FAF0-1FAF8, U+1FB00-1FBFF;
+}
+/* vietnamese */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmbiArmlw.woff2) format('woff2');
+ unicode-range: U+0102-0103, U+0110-0111, U+0128-0129, U+0168-0169, U+01A0-01A1, U+01AF-01B0, U+0300-0301, U+0303-0304, U+0308-0309, U+0323, U+0329, U+1EA0-1EF9, U+20AB;
+}
+/* latin-ext */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmaiArmlw.woff2) format('woff2');
+ unicode-range: U+0100-02BA, U+02BD-02C5, U+02C7-02CC, U+02CE-02D7, U+02DD-02FF, U+0304, U+0308, U+0329, U+1D00-1DBF, U+1E00-1E9F, U+1EF2-1EFF, U+2020, U+20A0-20AB, U+20AD-20C0, U+2113, U+2C60-2C7F, U+A720-A7FF;
+}
+/* latin */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmUiAo.woff2) format('woff2');
+ unicode-range: U+0000-00FF, U+0131, U+0152-0153, U+02BB-02BC, U+02C6, U+02DA, U+02DC, U+0304, U+0308, U+0329, U+2000-206F, U+20AC, U+2122, U+2191, U+2193, U+2212, U+2215, U+FEFF, U+FFFD;
+}
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmQiArmlw.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmQiArmlw.woff2
new file mode 100644
index 0000000..b0ed6d6
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmQiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmUiAo.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmUiAo.woff2
new file mode 100644
index 0000000..5a5fad1
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmUiAo.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmXiArmlw.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmXiArmlw.woff2
new file mode 100644
index 0000000..de646f8
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmXiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmYiArmlw.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmYiArmlw.woff2
new file mode 100644
index 0000000..6a6fb7a
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmYiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmZiArmlw.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmZiArmlw.woff2
new file mode 100644
index 0000000..1a1ed7c
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmZiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmaiArmlw.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmaiArmlw.woff2
new file mode 100644
index 0000000..47e69cf
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmaiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmbiArmlw.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmbiArmlw.woff2
new file mode 100644
index 0000000..bc95855
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmbiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVn6iArmlw.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVn6iArmlw.woff2
new file mode 100644
index 0000000..f9c26fa
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVn6iArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVnoiArmlw.woff2 b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVnoiArmlw.woff2
new file mode 100644
index 0000000..15e1583
Binary files /dev/null and b/docs/deps/Roboto-0.4.9/KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVnoiArmlw.woff2 differ
diff --git a/docs/deps/Roboto-0.4.9/font.css b/docs/deps/Roboto-0.4.9/font.css
new file mode 100644
index 0000000..33cb5f4
--- /dev/null
+++ b/docs/deps/Roboto-0.4.9/font.css
@@ -0,0 +1,90 @@
+/* cyrillic-ext */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmZiArmlw.woff2) format('woff2');
+ unicode-range: U+0460-052F, U+1C80-1C8A, U+20B4, U+2DE0-2DFF, U+A640-A69F, U+FE2E-FE2F;
+}
+/* cyrillic */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmQiArmlw.woff2) format('woff2');
+ unicode-range: U+0301, U+0400-045F, U+0490-0491, U+04B0-04B1, U+2116;
+}
+/* greek-ext */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmYiArmlw.woff2) format('woff2');
+ unicode-range: U+1F00-1FFF;
+}
+/* greek */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmXiArmlw.woff2) format('woff2');
+ unicode-range: U+0370-0377, U+037A-037F, U+0384-038A, U+038C, U+038E-03A1, U+03A3-03FF;
+}
+/* math */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVnoiArmlw.woff2) format('woff2');
+ unicode-range: U+0302-0303, U+0305, U+0307-0308, U+0310, U+0312, U+0315, U+031A, U+0326-0327, U+032C, U+032F-0330, U+0332-0333, U+0338, U+033A, U+0346, U+034D, U+0391-03A1, U+03A3-03A9, U+03B1-03C9, U+03D1, U+03D5-03D6, U+03F0-03F1, U+03F4-03F5, U+2016-2017, U+2034-2038, U+203C, U+2040, U+2043, U+2047, U+2050, U+2057, U+205F, U+2070-2071, U+2074-208E, U+2090-209C, U+20D0-20DC, U+20E1, U+20E5-20EF, U+2100-2112, U+2114-2115, U+2117-2121, U+2123-214F, U+2190, U+2192, U+2194-21AE, U+21B0-21E5, U+21F1-21F2, U+21F4-2211, U+2213-2214, U+2216-22FF, U+2308-230B, U+2310, U+2319, U+231C-2321, U+2336-237A, U+237C, U+2395, U+239B-23B7, U+23D0, U+23DC-23E1, U+2474-2475, U+25AF, U+25B3, U+25B7, U+25BD, U+25C1, U+25CA, U+25CC, U+25FB, U+266D-266F, U+27C0-27FF, U+2900-2AFF, U+2B0E-2B11, U+2B30-2B4C, U+2BFE, U+3030, U+FF5B, U+FF5D, U+1D400-1D7FF, U+1EE00-1EEFF;
+}
+/* symbols */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVn6iArmlw.woff2) format('woff2');
+ unicode-range: U+0001-000C, U+000E-001F, U+007F-009F, U+20DD-20E0, U+20E2-20E4, U+2150-218F, U+2190, U+2192, U+2194-2199, U+21AF, U+21E6-21F0, U+21F3, U+2218-2219, U+2299, U+22C4-22C6, U+2300-243F, U+2440-244A, U+2460-24FF, U+25A0-27BF, U+2800-28FF, U+2921-2922, U+2981, U+29BF, U+29EB, U+2B00-2BFF, U+4DC0-4DFF, U+FFF9-FFFB, U+10140-1018E, U+10190-1019C, U+101A0, U+101D0-101FD, U+102E0-102FB, U+10E60-10E7E, U+1D2C0-1D2D3, U+1D2E0-1D37F, U+1F000-1F0FF, U+1F100-1F1AD, U+1F1E6-1F1FF, U+1F30D-1F30F, U+1F315, U+1F31C, U+1F31E, U+1F320-1F32C, U+1F336, U+1F378, U+1F37D, U+1F382, U+1F393-1F39F, U+1F3A7-1F3A8, U+1F3AC-1F3AF, U+1F3C2, U+1F3C4-1F3C6, U+1F3CA-1F3CE, U+1F3D4-1F3E0, U+1F3ED, U+1F3F1-1F3F3, U+1F3F5-1F3F7, U+1F408, U+1F415, U+1F41F, U+1F426, U+1F43F, U+1F441-1F442, U+1F444, U+1F446-1F449, U+1F44C-1F44E, U+1F453, U+1F46A, U+1F47D, U+1F4A3, U+1F4B0, U+1F4B3, U+1F4B9, U+1F4BB, U+1F4BF, U+1F4C8-1F4CB, U+1F4D6, U+1F4DA, U+1F4DF, U+1F4E3-1F4E6, U+1F4EA-1F4ED, U+1F4F7, U+1F4F9-1F4FB, U+1F4FD-1F4FE, U+1F503, U+1F507-1F50B, U+1F50D, U+1F512-1F513, U+1F53E-1F54A, U+1F54F-1F5FA, U+1F610, U+1F650-1F67F, U+1F687, U+1F68D, U+1F691, U+1F694, U+1F698, U+1F6AD, U+1F6B2, U+1F6B9-1F6BA, U+1F6BC, U+1F6C6-1F6CF, U+1F6D3-1F6D7, U+1F6E0-1F6EA, U+1F6F0-1F6F3, U+1F6F7-1F6FC, U+1F700-1F7FF, U+1F800-1F80B, U+1F810-1F847, U+1F850-1F859, U+1F860-1F887, U+1F890-1F8AD, U+1F8B0-1F8BB, U+1F8C0-1F8C1, U+1F900-1F90B, U+1F93B, U+1F946, U+1F984, U+1F996, U+1F9E9, U+1FA00-1FA6F, U+1FA70-1FA7C, U+1FA80-1FA89, U+1FA8F-1FAC6, U+1FACE-1FADC, U+1FADF-1FAE9, U+1FAF0-1FAF8, U+1FB00-1FBFF;
+}
+/* vietnamese */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmbiArmlw.woff2) format('woff2');
+ unicode-range: U+0102-0103, U+0110-0111, U+0128-0129, U+0168-0169, U+01A0-01A1, U+01AF-01B0, U+0300-0301, U+0303-0304, U+0308-0309, U+0323, U+0329, U+1EA0-1EF9, U+20AB;
+}
+/* latin-ext */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmaiArmlw.woff2) format('woff2');
+ unicode-range: U+0100-02BA, U+02BD-02C5, U+02C7-02CC, U+02CE-02D7, U+02DD-02FF, U+0304, U+0308, U+0329, U+1D00-1DBF, U+1E00-1E9F, U+1EF2-1EFF, U+2020, U+20A0-20AB, U+20AD-20C0, U+2113, U+2C60-2C7F, U+A720-A7FF;
+}
+/* latin */
+@font-face {
+ font-family: 'Roboto';
+ font-style: normal;
+ font-weight: 400;
+ font-stretch: 100%;
+ font-display: swap;
+ src: url(KFOMCnqEu92Fr1ME7kSn66aGLdTylUAMQXC89YmC2DPNWubEbVmUiAo.woff2) format('woff2');
+ unicode-range: U+0000-00FF, U+0131, U+0152-0153, U+02BB-02BC, U+02C6, U+02DA, U+02DC, U+0304, U+0308, U+0329, U+2000-206F, U+20AC, U+2122, U+2191, U+2193, U+2212, U+2215, U+FEFF, U+FFFD;
+}
diff --git a/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_QOW4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_QOW4Ep0.woff2
new file mode 100644
index 0000000..db3b731
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_QOW4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_R-W4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_R-W4Ep0.woff2
new file mode 100644
index 0000000..f71b508
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_R-W4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_ROW4.woff2 b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_ROW4.woff2
new file mode 100644
index 0000000..6008f02
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_ROW4.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_S-W4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_S-W4Ep0.woff2
new file mode 100644
index 0000000..a7b593f
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_S-W4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SeW4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SeW4Ep0.woff2
new file mode 100644
index 0000000..5eca4ab
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SeW4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SuW4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SuW4Ep0.woff2
new file mode 100644
index 0000000..08e21fa
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.10/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SuW4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.10/font.css b/docs/deps/Roboto_Mono-0.4.10/font.css
new file mode 100644
index 0000000..fad8c87
--- /dev/null
+++ b/docs/deps/Roboto_Mono-0.4.10/font.css
@@ -0,0 +1,54 @@
+/* cyrillic-ext */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SeW4Ep0.woff2) format('woff2');
+ unicode-range: U+0460-052F, U+1C80-1C8A, U+20B4, U+2DE0-2DFF, U+A640-A69F, U+FE2E-FE2F;
+}
+/* cyrillic */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_QOW4Ep0.woff2) format('woff2');
+ unicode-range: U+0301, U+0400-045F, U+0490-0491, U+04B0-04B1, U+2116;
+}
+/* greek */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_R-W4Ep0.woff2) format('woff2');
+ unicode-range: U+0370-0377, U+037A-037F, U+0384-038A, U+038C, U+038E-03A1, U+03A3-03FF;
+}
+/* vietnamese */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_S-W4Ep0.woff2) format('woff2');
+ unicode-range: U+0102-0103, U+0110-0111, U+0128-0129, U+0168-0169, U+01A0-01A1, U+01AF-01B0, U+0300-0301, U+0303-0304, U+0308-0309, U+0323, U+0329, U+1EA0-1EF9, U+20AB;
+}
+/* latin-ext */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SuW4Ep0.woff2) format('woff2');
+ unicode-range: U+0100-02BA, U+02BD-02C5, U+02C7-02CC, U+02CE-02D7, U+02DD-02FF, U+0304, U+0308, U+0329, U+1D00-1DBF, U+1E00-1E9F, U+1EF2-1EFF, U+2020, U+20A0-20AB, U+20AD-20C0, U+2113, U+2C60-2C7F, U+A720-A7FF;
+}
+/* latin */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_ROW4.woff2) format('woff2');
+ unicode-range: U+0000-00FF, U+0131, U+0152-0153, U+02BB-02BC, U+02C6, U+02DA, U+02DC, U+0304, U+0308, U+0329, U+2000-206F, U+20AC, U+2122, U+2191, U+2193, U+2212, U+2215, U+FEFF, U+FFFD;
+}
diff --git a/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_QOW4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_QOW4Ep0.woff2
new file mode 100644
index 0000000..db3b731
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_QOW4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_R-W4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_R-W4Ep0.woff2
new file mode 100644
index 0000000..f71b508
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_R-W4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_ROW4.woff2 b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_ROW4.woff2
new file mode 100644
index 0000000..6008f02
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_ROW4.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_S-W4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_S-W4Ep0.woff2
new file mode 100644
index 0000000..a7b593f
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_S-W4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SeW4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SeW4Ep0.woff2
new file mode 100644
index 0000000..5eca4ab
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SeW4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SuW4Ep0.woff2 b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SuW4Ep0.woff2
new file mode 100644
index 0000000..08e21fa
Binary files /dev/null and b/docs/deps/Roboto_Mono-0.4.9/L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SuW4Ep0.woff2 differ
diff --git a/docs/deps/Roboto_Mono-0.4.9/font.css b/docs/deps/Roboto_Mono-0.4.9/font.css
new file mode 100644
index 0000000..fad8c87
--- /dev/null
+++ b/docs/deps/Roboto_Mono-0.4.9/font.css
@@ -0,0 +1,54 @@
+/* cyrillic-ext */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SeW4Ep0.woff2) format('woff2');
+ unicode-range: U+0460-052F, U+1C80-1C8A, U+20B4, U+2DE0-2DFF, U+A640-A69F, U+FE2E-FE2F;
+}
+/* cyrillic */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_QOW4Ep0.woff2) format('woff2');
+ unicode-range: U+0301, U+0400-045F, U+0490-0491, U+04B0-04B1, U+2116;
+}
+/* greek */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_R-W4Ep0.woff2) format('woff2');
+ unicode-range: U+0370-0377, U+037A-037F, U+0384-038A, U+038C, U+038E-03A1, U+03A3-03FF;
+}
+/* vietnamese */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_S-W4Ep0.woff2) format('woff2');
+ unicode-range: U+0102-0103, U+0110-0111, U+0128-0129, U+0168-0169, U+01A0-01A1, U+01AF-01B0, U+0300-0301, U+0303-0304, U+0308-0309, U+0323, U+0329, U+1EA0-1EF9, U+20AB;
+}
+/* latin-ext */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_SuW4Ep0.woff2) format('woff2');
+ unicode-range: U+0100-02BA, U+02BD-02C5, U+02C7-02CC, U+02CE-02D7, U+02DD-02FF, U+0304, U+0308, U+0329, U+1D00-1DBF, U+1E00-1E9F, U+1EF2-1EFF, U+2020, U+20A0-20AB, U+20AD-20C0, U+2113, U+2C60-2C7F, U+A720-A7FF;
+}
+/* latin */
+@font-face {
+ font-family: 'Roboto Mono';
+ font-style: normal;
+ font-weight: 400;
+ font-display: swap;
+ src: url(L0xuDF4xlVMF-BfR8bXMIhJHg45mwgGEFl0_3vq_ROW4.woff2) format('woff2');
+ unicode-range: U+0000-00FF, U+0131, U+0152-0153, U+02BB-02BC, U+02C6, U+02DA, U+02DC, U+0304, U+0308, U+0329, U+2000-206F, U+20AC, U+2122, U+2191, U+2193, U+2212, U+2215, U+FEFF, U+FFFD;
+}
diff --git a/docs/deps/bootstrap-5.3.1/bootstrap.bundle.min.js b/docs/deps/bootstrap-5.3.1/bootstrap.bundle.min.js
new file mode 100644
index 0000000..e8f21f7
--- /dev/null
+++ b/docs/deps/bootstrap-5.3.1/bootstrap.bundle.min.js
@@ -0,0 +1,7 @@
+/*!
+ * Bootstrap v5.3.1 (https://getbootstrap.com/)
+ * Copyright 2011-2023 The Bootstrap Authors (https://github.com/twbs/bootstrap/graphs/contributors)
+ * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)
+ */
+!function(t,e){"object"==typeof exports&&"undefined"!=typeof module?module.exports=e():"function"==typeof define&&define.amd?define(e):(t="undefined"!=typeof globalThis?globalThis:t||self).bootstrap=e()}(this,(function(){"use strict";const t=new Map,e={set(e,i,n){t.has(e)||t.set(e,new Map);const s=t.get(e);s.has(i)||0===s.size?s.set(i,n):console.error(`Bootstrap doesn't allow more than one instance per element. Bound instance: ${Array.from(s.keys())[0]}.`)},get:(e,i)=>t.has(e)&&t.get(e).get(i)||null,remove(e,i){if(!t.has(e))return;const n=t.get(e);n.delete(i),0===n.size&&t.delete(e)}},i="transitionend",n=t=>(t&&window.CSS&&window.CSS.escape&&(t=t.replace(/#([^\s"#']+)/g,((t,e)=>`#${CSS.escape(e)}`))),t),s=t=>{t.dispatchEvent(new Event(i))},o=t=>!(!t||"object"!=typeof t)&&(void 0!==t.jquery&&(t=t[0]),void 0!==t.nodeType),r=t=>o(t)?t.jquery?t[0]:t:"string"==typeof t&&t.length>0?document.querySelector(n(t)):null,a=t=>{if(!o(t)||0===t.getClientRects().length)return!1;const e="visible"===getComputedStyle(t).getPropertyValue("visibility"),i=t.closest("details:not([open])");if(!i)return e;if(i!==t){const e=t.closest("summary");if(e&&e.parentNode!==i)return!1;if(null===e)return!1}return e},l=t=>!t||t.nodeType!==Node.ELEMENT_NODE||!!t.classList.contains("disabled")||(void 0!==t.disabled?t.disabled:t.hasAttribute("disabled")&&"false"!==t.getAttribute("disabled")),c=t=>{if(!document.documentElement.attachShadow)return null;if("function"==typeof t.getRootNode){const e=t.getRootNode();return e instanceof ShadowRoot?e:null}return t instanceof ShadowRoot?t:t.parentNode?c(t.parentNode):null},h=()=>{},d=t=>{t.offsetHeight},u=()=>window.jQuery&&!document.body.hasAttribute("data-bs-no-jquery")?window.jQuery:null,f=[],p=()=>"rtl"===document.documentElement.dir,m=t=>{var e;e=()=>{const e=u();if(e){const i=t.NAME,n=e.fn[i];e.fn[i]=t.jQueryInterface,e.fn[i].Constructor=t,e.fn[i].noConflict=()=>(e.fn[i]=n,t.jQueryInterface)}},"loading"===document.readyState?(f.length||document.addEventListener("DOMContentLoaded",(()=>{for(const t of f)t()})),f.push(e)):e()},g=(t,e=[],i=t)=>"function"==typeof t?t(...e):i,_=(t,e,n=!0)=>{if(!n)return void g(t);const o=(t=>{if(!t)return 0;let{transitionDuration:e,transitionDelay:i}=window.getComputedStyle(t);const n=Number.parseFloat(e),s=Number.parseFloat(i);return n||s?(e=e.split(",")[0],i=i.split(",")[0],1e3*(Number.parseFloat(e)+Number.parseFloat(i))):0})(e)+5;let r=!1;const a=({target:n})=>{n===e&&(r=!0,e.removeEventListener(i,a),g(t))};e.addEventListener(i,a),setTimeout((()=>{r||s(e)}),o)},b=(t,e,i,n)=>{const s=t.length;let o=t.indexOf(e);return-1===o?!i&&n?t[s-1]:t[0]:(o+=i?1:-1,n&&(o=(o+s)%s),t[Math.max(0,Math.min(o,s-1))])},v=/[^.]*(?=\..*)\.|.*/,y=/\..*/,w=/::\d+$/,A={};let E=1;const T={mouseenter:"mouseover",mouseleave:"mouseout"},C=new Set(["click","dblclick","mouseup","mousedown","contextmenu","mousewheel","DOMMouseScroll","mouseover","mouseout","mousemove","selectstart","selectend","keydown","keypress","keyup","orientationchange","touchstart","touchmove","touchend","touchcancel","pointerdown","pointermove","pointerup","pointerleave","pointercancel","gesturestart","gesturechange","gestureend","focus","blur","change","reset","select","submit","focusin","focusout","load","unload","beforeunload","resize","move","DOMContentLoaded","readystatechange","error","abort","scroll"]);function O(t,e){return e&&`${e}::${E++}`||t.uidEvent||E++}function x(t){const e=O(t);return t.uidEvent=e,A[e]=A[e]||{},A[e]}function k(t,e,i=null){return Object.values(t).find((t=>t.callable===e&&t.delegationSelector===i))}function L(t,e,i){const n="string"==typeof e,s=n?i:e||i;let o=I(t);return C.has(o)||(o=t),[n,s,o]}function S(t,e,i,n,s){if("string"!=typeof e||!t)return;let[o,r,a]=L(e,i,n);if(e in T){const t=t=>function(e){if(!e.relatedTarget||e.relatedTarget!==e.delegateTarget&&!e.delegateTarget.contains(e.relatedTarget))return t.call(this,e)};r=t(r)}const l=x(t),c=l[a]||(l[a]={}),h=k(c,r,o?i:null);if(h)return void(h.oneOff=h.oneOff&&s);const d=O(r,e.replace(v,"")),u=o?function(t,e,i){return function n(s){const o=t.querySelectorAll(e);for(let{target:r}=s;r&&r!==this;r=r.parentNode)for(const a of o)if(a===r)return P(s,{delegateTarget:r}),n.oneOff&&N.off(t,s.type,e,i),i.apply(r,[s])}}(t,i,r):function(t,e){return function i(n){return P(n,{delegateTarget:t}),i.oneOff&&N.off(t,n.type,e),e.apply(t,[n])}}(t,r);u.delegationSelector=o?i:null,u.callable=r,u.oneOff=s,u.uidEvent=d,c[d]=u,t.addEventListener(a,u,o)}function D(t,e,i,n,s){const o=k(e[i],n,s);o&&(t.removeEventListener(i,o,Boolean(s)),delete e[i][o.uidEvent])}function $(t,e,i,n){const s=e[i]||{};for(const[o,r]of Object.entries(s))o.includes(n)&&D(t,e,i,r.callable,r.delegationSelector)}function I(t){return t=t.replace(y,""),T[t]||t}const N={on(t,e,i,n){S(t,e,i,n,!1)},one(t,e,i,n){S(t,e,i,n,!0)},off(t,e,i,n){if("string"!=typeof e||!t)return;const[s,o,r]=L(e,i,n),a=r!==e,l=x(t),c=l[r]||{},h=e.startsWith(".");if(void 0===o){if(h)for(const i of Object.keys(l))$(t,l,i,e.slice(1));for(const[i,n]of Object.entries(c)){const s=i.replace(w,"");a&&!e.includes(s)||D(t,l,r,n.callable,n.delegationSelector)}}else{if(!Object.keys(c).length)return;D(t,l,r,o,s?i:null)}},trigger(t,e,i){if("string"!=typeof e||!t)return null;const n=u();let s=null,o=!0,r=!0,a=!1;e!==I(e)&&n&&(s=n.Event(e,i),n(t).trigger(s),o=!s.isPropagationStopped(),r=!s.isImmediatePropagationStopped(),a=s.isDefaultPrevented());const l=P(new Event(e,{bubbles:o,cancelable:!0}),i);return a&&l.preventDefault(),r&&t.dispatchEvent(l),l.defaultPrevented&&s&&s.preventDefault(),l}};function P(t,e={}){for(const[i,n]of Object.entries(e))try{t[i]=n}catch(e){Object.defineProperty(t,i,{configurable:!0,get:()=>n})}return t}function M(t){if("true"===t)return!0;if("false"===t)return!1;if(t===Number(t).toString())return Number(t);if(""===t||"null"===t)return null;if("string"!=typeof t)return t;try{return JSON.parse(decodeURIComponent(t))}catch(e){return t}}function j(t){return t.replace(/[A-Z]/g,(t=>`-${t.toLowerCase()}`))}const F={setDataAttribute(t,e,i){t.setAttribute(`data-bs-${j(e)}`,i)},removeDataAttribute(t,e){t.removeAttribute(`data-bs-${j(e)}`)},getDataAttributes(t){if(!t)return{};const e={},i=Object.keys(t.dataset).filter((t=>t.startsWith("bs")&&!t.startsWith("bsConfig")));for(const n of i){let i=n.replace(/^bs/,"");i=i.charAt(0).toLowerCase()+i.slice(1,i.length),e[i]=M(t.dataset[n])}return e},getDataAttribute:(t,e)=>M(t.getAttribute(`data-bs-${j(e)}`))};class H{static get Default(){return{}}static get DefaultType(){return{}}static get NAME(){throw new Error('You have to implement the static method "NAME", for each component!')}_getConfig(t){return t=this._mergeConfigObj(t),t=this._configAfterMerge(t),this._typeCheckConfig(t),t}_configAfterMerge(t){return t}_mergeConfigObj(t,e){const i=o(e)?F.getDataAttribute(e,"config"):{};return{...this.constructor.Default,..."object"==typeof i?i:{},...o(e)?F.getDataAttributes(e):{},..."object"==typeof t?t:{}}}_typeCheckConfig(t,e=this.constructor.DefaultType){for(const[n,s]of Object.entries(e)){const e=t[n],r=o(e)?"element":null==(i=e)?`${i}`:Object.prototype.toString.call(i).match(/\s([a-z]+)/i)[1].toLowerCase();if(!new RegExp(s).test(r))throw new TypeError(`${this.constructor.NAME.toUpperCase()}: Option "${n}" provided type "${r}" but expected type "${s}".`)}var i}}class W extends H{constructor(t,i){super(),(t=r(t))&&(this._element=t,this._config=this._getConfig(i),e.set(this._element,this.constructor.DATA_KEY,this))}dispose(){e.remove(this._element,this.constructor.DATA_KEY),N.off(this._element,this.constructor.EVENT_KEY);for(const t of Object.getOwnPropertyNames(this))this[t]=null}_queueCallback(t,e,i=!0){_(t,e,i)}_getConfig(t){return t=this._mergeConfigObj(t,this._element),t=this._configAfterMerge(t),this._typeCheckConfig(t),t}static getInstance(t){return e.get(r(t),this.DATA_KEY)}static getOrCreateInstance(t,e={}){return this.getInstance(t)||new this(t,"object"==typeof e?e:null)}static get VERSION(){return"5.3.1"}static get DATA_KEY(){return`bs.${this.NAME}`}static get EVENT_KEY(){return`.${this.DATA_KEY}`}static eventName(t){return`${t}${this.EVENT_KEY}`}}const B=t=>{let e=t.getAttribute("data-bs-target");if(!e||"#"===e){let i=t.getAttribute("href");if(!i||!i.includes("#")&&!i.startsWith("."))return null;i.includes("#")&&!i.startsWith("#")&&(i=`#${i.split("#")[1]}`),e=i&&"#"!==i?i.trim():null}return n(e)},z={find:(t,e=document.documentElement)=>[].concat(...Element.prototype.querySelectorAll.call(e,t)),findOne:(t,e=document.documentElement)=>Element.prototype.querySelector.call(e,t),children:(t,e)=>[].concat(...t.children).filter((t=>t.matches(e))),parents(t,e){const i=[];let n=t.parentNode.closest(e);for(;n;)i.push(n),n=n.parentNode.closest(e);return i},prev(t,e){let i=t.previousElementSibling;for(;i;){if(i.matches(e))return[i];i=i.previousElementSibling}return[]},next(t,e){let i=t.nextElementSibling;for(;i;){if(i.matches(e))return[i];i=i.nextElementSibling}return[]},focusableChildren(t){const e=["a","button","input","textarea","select","details","[tabindex]",'[contenteditable="true"]'].map((t=>`${t}:not([tabindex^="-"])`)).join(",");return this.find(e,t).filter((t=>!l(t)&&a(t)))},getSelectorFromElement(t){const e=B(t);return e&&z.findOne(e)?e:null},getElementFromSelector(t){const e=B(t);return e?z.findOne(e):null},getMultipleElementsFromSelector(t){const e=B(t);return e?z.find(e):[]}},R=(t,e="hide")=>{const i=`click.dismiss${t.EVENT_KEY}`,n=t.NAME;N.on(document,i,`[data-bs-dismiss="${n}"]`,(function(i){if(["A","AREA"].includes(this.tagName)&&i.preventDefault(),l(this))return;const s=z.getElementFromSelector(this)||this.closest(`.${n}`);t.getOrCreateInstance(s)[e]()}))},q=".bs.alert",V=`close${q}`,K=`closed${q}`;class Q extends W{static get NAME(){return"alert"}close(){if(N.trigger(this._element,V).defaultPrevented)return;this._element.classList.remove("show");const t=this._element.classList.contains("fade");this._queueCallback((()=>this._destroyElement()),this._element,t)}_destroyElement(){this._element.remove(),N.trigger(this._element,K),this.dispose()}static jQueryInterface(t){return this.each((function(){const e=Q.getOrCreateInstance(this);if("string"==typeof t){if(void 0===e[t]||t.startsWith("_")||"constructor"===t)throw new TypeError(`No method named "${t}"`);e[t](this)}}))}}R(Q,"close"),m(Q);const X='[data-bs-toggle="button"]';class Y extends W{static get NAME(){return"button"}toggle(){this._element.setAttribute("aria-pressed",this._element.classList.toggle("active"))}static jQueryInterface(t){return this.each((function(){const e=Y.getOrCreateInstance(this);"toggle"===t&&e[t]()}))}}N.on(document,"click.bs.button.data-api",X,(t=>{t.preventDefault();const e=t.target.closest(X);Y.getOrCreateInstance(e).toggle()})),m(Y);const U=".bs.swipe",G=`touchstart${U}`,J=`touchmove${U}`,Z=`touchend${U}`,tt=`pointerdown${U}`,et=`pointerup${U}`,it={endCallback:null,leftCallback:null,rightCallback:null},nt={endCallback:"(function|null)",leftCallback:"(function|null)",rightCallback:"(function|null)"};class st extends H{constructor(t,e){super(),this._element=t,t&&st.isSupported()&&(this._config=this._getConfig(e),this._deltaX=0,this._supportPointerEvents=Boolean(window.PointerEvent),this._initEvents())}static get Default(){return it}static get DefaultType(){return nt}static get NAME(){return"swipe"}dispose(){N.off(this._element,U)}_start(t){this._supportPointerEvents?this._eventIsPointerPenTouch(t)&&(this._deltaX=t.clientX):this._deltaX=t.touches[0].clientX}_end(t){this._eventIsPointerPenTouch(t)&&(this._deltaX=t.clientX-this._deltaX),this._handleSwipe(),g(this._config.endCallback)}_move(t){this._deltaX=t.touches&&t.touches.length>1?0:t.touches[0].clientX-this._deltaX}_handleSwipe(){const t=Math.abs(this._deltaX);if(t<=40)return;const e=t/this._deltaX;this._deltaX=0,e&&g(e>0?this._config.rightCallback:this._config.leftCallback)}_initEvents(){this._supportPointerEvents?(N.on(this._element,tt,(t=>this._start(t))),N.on(this._element,et,(t=>this._end(t))),this._element.classList.add("pointer-event")):(N.on(this._element,G,(t=>this._start(t))),N.on(this._element,J,(t=>this._move(t))),N.on(this._element,Z,(t=>this._end(t))))}_eventIsPointerPenTouch(t){return this._supportPointerEvents&&("pen"===t.pointerType||"touch"===t.pointerType)}static isSupported(){return"ontouchstart"in document.documentElement||navigator.maxTouchPoints>0}}const ot=".bs.carousel",rt=".data-api",at="next",lt="prev",ct="left",ht="right",dt=`slide${ot}`,ut=`slid${ot}`,ft=`keydown${ot}`,pt=`mouseenter${ot}`,mt=`mouseleave${ot}`,gt=`dragstart${ot}`,_t=`load${ot}${rt}`,bt=`click${ot}${rt}`,vt="carousel",yt="active",wt=".active",At=".carousel-item",Et=wt+At,Tt={ArrowLeft:ht,ArrowRight:ct},Ct={interval:5e3,keyboard:!0,pause:"hover",ride:!1,touch:!0,wrap:!0},Ot={interval:"(number|boolean)",keyboard:"boolean",pause:"(string|boolean)",ride:"(boolean|string)",touch:"boolean",wrap:"boolean"};class xt extends W{constructor(t,e){super(t,e),this._interval=null,this._activeElement=null,this._isSliding=!1,this.touchTimeout=null,this._swipeHelper=null,this._indicatorsElement=z.findOne(".carousel-indicators",this._element),this._addEventListeners(),this._config.ride===vt&&this.cycle()}static get Default(){return Ct}static get DefaultType(){return Ot}static get NAME(){return"carousel"}next(){this._slide(at)}nextWhenVisible(){!document.hidden&&a(this._element)&&this.next()}prev(){this._slide(lt)}pause(){this._isSliding&&s(this._element),this._clearInterval()}cycle(){this._clearInterval(),this._updateInterval(),this._interval=setInterval((()=>this.nextWhenVisible()),this._config.interval)}_maybeEnableCycle(){this._config.ride&&(this._isSliding?N.one(this._element,ut,(()=>this.cycle())):this.cycle())}to(t){const e=this._getItems();if(t>e.length-1||t<0)return;if(this._isSliding)return void N.one(this._element,ut,(()=>this.to(t)));const i=this._getItemIndex(this._getActive());if(i===t)return;const n=t>i?at:lt;this._slide(n,e[t])}dispose(){this._swipeHelper&&this._swipeHelper.dispose(),super.dispose()}_configAfterMerge(t){return t.defaultInterval=t.interval,t}_addEventListeners(){this._config.keyboard&&N.on(this._element,ft,(t=>this._keydown(t))),"hover"===this._config.pause&&(N.on(this._element,pt,(()=>this.pause())),N.on(this._element,mt,(()=>this._maybeEnableCycle()))),this._config.touch&&st.isSupported()&&this._addTouchEventListeners()}_addTouchEventListeners(){for(const t of z.find(".carousel-item img",this._element))N.on(t,gt,(t=>t.preventDefault()));const t={leftCallback:()=>this._slide(this._directionToOrder(ct)),rightCallback:()=>this._slide(this._directionToOrder(ht)),endCallback:()=>{"hover"===this._config.pause&&(this.pause(),this.touchTimeout&&clearTimeout(this.touchTimeout),this.touchTimeout=setTimeout((()=>this._maybeEnableCycle()),500+this._config.interval))}};this._swipeHelper=new st(this._element,t)}_keydown(t){if(/input|textarea/i.test(t.target.tagName))return;const e=Tt[t.key];e&&(t.preventDefault(),this._slide(this._directionToOrder(e)))}_getItemIndex(t){return this._getItems().indexOf(t)}_setActiveIndicatorElement(t){if(!this._indicatorsElement)return;const e=z.findOne(wt,this._indicatorsElement);e.classList.remove(yt),e.removeAttribute("aria-current");const i=z.findOne(`[data-bs-slide-to="${t}"]`,this._indicatorsElement);i&&(i.classList.add(yt),i.setAttribute("aria-current","true"))}_updateInterval(){const t=this._activeElement||this._getActive();if(!t)return;const e=Number.parseInt(t.getAttribute("data-bs-interval"),10);this._config.interval=e||this._config.defaultInterval}_slide(t,e=null){if(this._isSliding)return;const i=this._getActive(),n=t===at,s=e||b(this._getItems(),i,n,this._config.wrap);if(s===i)return;const o=this._getItemIndex(s),r=e=>N.trigger(this._element,e,{relatedTarget:s,direction:this._orderToDirection(t),from:this._getItemIndex(i),to:o});if(r(dt).defaultPrevented)return;if(!i||!s)return;const a=Boolean(this._interval);this.pause(),this._isSliding=!0,this._setActiveIndicatorElement(o),this._activeElement=s;const l=n?"carousel-item-start":"carousel-item-end",c=n?"carousel-item-next":"carousel-item-prev";s.classList.add(c),d(s),i.classList.add(l),s.classList.add(l),this._queueCallback((()=>{s.classList.remove(l,c),s.classList.add(yt),i.classList.remove(yt,c,l),this._isSliding=!1,r(ut)}),i,this._isAnimated()),a&&this.cycle()}_isAnimated(){return this._element.classList.contains("slide")}_getActive(){return z.findOne(Et,this._element)}_getItems(){return z.find(At,this._element)}_clearInterval(){this._interval&&(clearInterval(this._interval),this._interval=null)}_directionToOrder(t){return p()?t===ct?lt:at:t===ct?at:lt}_orderToDirection(t){return p()?t===lt?ct:ht:t===lt?ht:ct}static jQueryInterface(t){return this.each((function(){const e=xt.getOrCreateInstance(this,t);if("number"!=typeof t){if("string"==typeof t){if(void 0===e[t]||t.startsWith("_")||"constructor"===t)throw new TypeError(`No method named "${t}"`);e[t]()}}else e.to(t)}))}}N.on(document,bt,"[data-bs-slide], [data-bs-slide-to]",(function(t){const e=z.getElementFromSelector(this);if(!e||!e.classList.contains(vt))return;t.preventDefault();const i=xt.getOrCreateInstance(e),n=this.getAttribute("data-bs-slide-to");return n?(i.to(n),void i._maybeEnableCycle()):"next"===F.getDataAttribute(this,"slide")?(i.next(),void i._maybeEnableCycle()):(i.prev(),void i._maybeEnableCycle())})),N.on(window,_t,(()=>{const t=z.find('[data-bs-ride="carousel"]');for(const e of t)xt.getOrCreateInstance(e)})),m(xt);const kt=".bs.collapse",Lt=`show${kt}`,St=`shown${kt}`,Dt=`hide${kt}`,$t=`hidden${kt}`,It=`click${kt}.data-api`,Nt="show",Pt="collapse",Mt="collapsing",jt=`:scope .${Pt} .${Pt}`,Ft='[data-bs-toggle="collapse"]',Ht={parent:null,toggle:!0},Wt={parent:"(null|element)",toggle:"boolean"};class Bt extends W{constructor(t,e){super(t,e),this._isTransitioning=!1,this._triggerArray=[];const i=z.find(Ft);for(const t of i){const e=z.getSelectorFromElement(t),i=z.find(e).filter((t=>t===this._element));null!==e&&i.length&&this._triggerArray.push(t)}this._initializeChildren(),this._config.parent||this._addAriaAndCollapsedClass(this._triggerArray,this._isShown()),this._config.toggle&&this.toggle()}static get Default(){return Ht}static get DefaultType(){return Wt}static get NAME(){return"collapse"}toggle(){this._isShown()?this.hide():this.show()}show(){if(this._isTransitioning||this._isShown())return;let t=[];if(this._config.parent&&(t=this._getFirstLevelChildren(".collapse.show, .collapse.collapsing").filter((t=>t!==this._element)).map((t=>Bt.getOrCreateInstance(t,{toggle:!1})))),t.length&&t[0]._isTransitioning)return;if(N.trigger(this._element,Lt).defaultPrevented)return;for(const e of t)e.hide();const e=this._getDimension();this._element.classList.remove(Pt),this._element.classList.add(Mt),this._element.style[e]=0,this._addAriaAndCollapsedClass(this._triggerArray,!0),this._isTransitioning=!0;const i=`scroll${e[0].toUpperCase()+e.slice(1)}`;this._queueCallback((()=>{this._isTransitioning=!1,this._element.classList.remove(Mt),this._element.classList.add(Pt,Nt),this._element.style[e]="",N.trigger(this._element,St)}),this._element,!0),this._element.style[e]=`${this._element[i]}px`}hide(){if(this._isTransitioning||!this._isShown())return;if(N.trigger(this._element,Dt).defaultPrevented)return;const t=this._getDimension();this._element.style[t]=`${this._element.getBoundingClientRect()[t]}px`,d(this._element),this._element.classList.add(Mt),this._element.classList.remove(Pt,Nt);for(const t of this._triggerArray){const e=z.getElementFromSelector(t);e&&!this._isShown(e)&&this._addAriaAndCollapsedClass([t],!1)}this._isTransitioning=!0,this._element.style[t]="",this._queueCallback((()=>{this._isTransitioning=!1,this._element.classList.remove(Mt),this._element.classList.add(Pt),N.trigger(this._element,$t)}),this._element,!0)}_isShown(t=this._element){return t.classList.contains(Nt)}_configAfterMerge(t){return t.toggle=Boolean(t.toggle),t.parent=r(t.parent),t}_getDimension(){return this._element.classList.contains("collapse-horizontal")?"width":"height"}_initializeChildren(){if(!this._config.parent)return;const t=this._getFirstLevelChildren(Ft);for(const e of t){const t=z.getElementFromSelector(e);t&&this._addAriaAndCollapsedClass([e],this._isShown(t))}}_getFirstLevelChildren(t){const e=z.find(jt,this._config.parent);return z.find(t,this._config.parent).filter((t=>!e.includes(t)))}_addAriaAndCollapsedClass(t,e){if(t.length)for(const i of t)i.classList.toggle("collapsed",!e),i.setAttribute("aria-expanded",e)}static jQueryInterface(t){const e={};return"string"==typeof t&&/show|hide/.test(t)&&(e.toggle=!1),this.each((function(){const i=Bt.getOrCreateInstance(this,e);if("string"==typeof t){if(void 0===i[t])throw new TypeError(`No method named "${t}"`);i[t]()}}))}}N.on(document,It,Ft,(function(t){("A"===t.target.tagName||t.delegateTarget&&"A"===t.delegateTarget.tagName)&&t.preventDefault();for(const t of z.getMultipleElementsFromSelector(this))Bt.getOrCreateInstance(t,{toggle:!1}).toggle()})),m(Bt);var zt="top",Rt="bottom",qt="right",Vt="left",Kt="auto",Qt=[zt,Rt,qt,Vt],Xt="start",Yt="end",Ut="clippingParents",Gt="viewport",Jt="popper",Zt="reference",te=Qt.reduce((function(t,e){return t.concat([e+"-"+Xt,e+"-"+Yt])}),[]),ee=[].concat(Qt,[Kt]).reduce((function(t,e){return t.concat([e,e+"-"+Xt,e+"-"+Yt])}),[]),ie="beforeRead",ne="read",se="afterRead",oe="beforeMain",re="main",ae="afterMain",le="beforeWrite",ce="write",he="afterWrite",de=[ie,ne,se,oe,re,ae,le,ce,he];function ue(t){return t?(t.nodeName||"").toLowerCase():null}function fe(t){if(null==t)return window;if("[object Window]"!==t.toString()){var e=t.ownerDocument;return e&&e.defaultView||window}return t}function pe(t){return t instanceof fe(t).Element||t instanceof Element}function me(t){return t instanceof fe(t).HTMLElement||t instanceof HTMLElement}function ge(t){return"undefined"!=typeof ShadowRoot&&(t instanceof fe(t).ShadowRoot||t instanceof ShadowRoot)}const _e={name:"applyStyles",enabled:!0,phase:"write",fn:function(t){var e=t.state;Object.keys(e.elements).forEach((function(t){var i=e.styles[t]||{},n=e.attributes[t]||{},s=e.elements[t];me(s)&&ue(s)&&(Object.assign(s.style,i),Object.keys(n).forEach((function(t){var e=n[t];!1===e?s.removeAttribute(t):s.setAttribute(t,!0===e?"":e)})))}))},effect:function(t){var e=t.state,i={popper:{position:e.options.strategy,left:"0",top:"0",margin:"0"},arrow:{position:"absolute"},reference:{}};return Object.assign(e.elements.popper.style,i.popper),e.styles=i,e.elements.arrow&&Object.assign(e.elements.arrow.style,i.arrow),function(){Object.keys(e.elements).forEach((function(t){var n=e.elements[t],s=e.attributes[t]||{},o=Object.keys(e.styles.hasOwnProperty(t)?e.styles[t]:i[t]).reduce((function(t,e){return t[e]="",t}),{});me(n)&&ue(n)&&(Object.assign(n.style,o),Object.keys(s).forEach((function(t){n.removeAttribute(t)})))}))}},requires:["computeStyles"]};function be(t){return t.split("-")[0]}var ve=Math.max,ye=Math.min,we=Math.round;function Ae(){var t=navigator.userAgentData;return null!=t&&t.brands&&Array.isArray(t.brands)?t.brands.map((function(t){return t.brand+"/"+t.version})).join(" "):navigator.userAgent}function Ee(){return!/^((?!chrome|android).)*safari/i.test(Ae())}function Te(t,e,i){void 0===e&&(e=!1),void 0===i&&(i=!1);var n=t.getBoundingClientRect(),s=1,o=1;e&&me(t)&&(s=t.offsetWidth>0&&we(n.width)/t.offsetWidth||1,o=t.offsetHeight>0&&we(n.height)/t.offsetHeight||1);var r=(pe(t)?fe(t):window).visualViewport,a=!Ee()&&i,l=(n.left+(a&&r?r.offsetLeft:0))/s,c=(n.top+(a&&r?r.offsetTop:0))/o,h=n.width/s,d=n.height/o;return{width:h,height:d,top:c,right:l+h,bottom:c+d,left:l,x:l,y:c}}function Ce(t){var e=Te(t),i=t.offsetWidth,n=t.offsetHeight;return Math.abs(e.width-i)<=1&&(i=e.width),Math.abs(e.height-n)<=1&&(n=e.height),{x:t.offsetLeft,y:t.offsetTop,width:i,height:n}}function Oe(t,e){var i=e.getRootNode&&e.getRootNode();if(t.contains(e))return!0;if(i&&ge(i)){var n=e;do{if(n&&t.isSameNode(n))return!0;n=n.parentNode||n.host}while(n)}return!1}function xe(t){return fe(t).getComputedStyle(t)}function ke(t){return["table","td","th"].indexOf(ue(t))>=0}function Le(t){return((pe(t)?t.ownerDocument:t.document)||window.document).documentElement}function Se(t){return"html"===ue(t)?t:t.assignedSlot||t.parentNode||(ge(t)?t.host:null)||Le(t)}function De(t){return me(t)&&"fixed"!==xe(t).position?t.offsetParent:null}function $e(t){for(var e=fe(t),i=De(t);i&&ke(i)&&"static"===xe(i).position;)i=De(i);return i&&("html"===ue(i)||"body"===ue(i)&&"static"===xe(i).position)?e:i||function(t){var e=/firefox/i.test(Ae());if(/Trident/i.test(Ae())&&me(t)&&"fixed"===xe(t).position)return null;var i=Se(t);for(ge(i)&&(i=i.host);me(i)&&["html","body"].indexOf(ue(i))<0;){var n=xe(i);if("none"!==n.transform||"none"!==n.perspective||"paint"===n.contain||-1!==["transform","perspective"].indexOf(n.willChange)||e&&"filter"===n.willChange||e&&n.filter&&"none"!==n.filter)return i;i=i.parentNode}return null}(t)||e}function Ie(t){return["top","bottom"].indexOf(t)>=0?"x":"y"}function Ne(t,e,i){return ve(t,ye(e,i))}function Pe(t){return Object.assign({},{top:0,right:0,bottom:0,left:0},t)}function Me(t,e){return e.reduce((function(e,i){return e[i]=t,e}),{})}const je={name:"arrow",enabled:!0,phase:"main",fn:function(t){var e,i=t.state,n=t.name,s=t.options,o=i.elements.arrow,r=i.modifiersData.popperOffsets,a=be(i.placement),l=Ie(a),c=[Vt,qt].indexOf(a)>=0?"height":"width";if(o&&r){var h=function(t,e){return Pe("number"!=typeof(t="function"==typeof t?t(Object.assign({},e.rects,{placement:e.placement})):t)?t:Me(t,Qt))}(s.padding,i),d=Ce(o),u="y"===l?zt:Vt,f="y"===l?Rt:qt,p=i.rects.reference[c]+i.rects.reference[l]-r[l]-i.rects.popper[c],m=r[l]-i.rects.reference[l],g=$e(o),_=g?"y"===l?g.clientHeight||0:g.clientWidth||0:0,b=p/2-m/2,v=h[u],y=_-d[c]-h[f],w=_/2-d[c]/2+b,A=Ne(v,w,y),E=l;i.modifiersData[n]=((e={})[E]=A,e.centerOffset=A-w,e)}},effect:function(t){var e=t.state,i=t.options.element,n=void 0===i?"[data-popper-arrow]":i;null!=n&&("string"!=typeof n||(n=e.elements.popper.querySelector(n)))&&Oe(e.elements.popper,n)&&(e.elements.arrow=n)},requires:["popperOffsets"],requiresIfExists:["preventOverflow"]};function Fe(t){return t.split("-")[1]}var He={top:"auto",right:"auto",bottom:"auto",left:"auto"};function We(t){var e,i=t.popper,n=t.popperRect,s=t.placement,o=t.variation,r=t.offsets,a=t.position,l=t.gpuAcceleration,c=t.adaptive,h=t.roundOffsets,d=t.isFixed,u=r.x,f=void 0===u?0:u,p=r.y,m=void 0===p?0:p,g="function"==typeof h?h({x:f,y:m}):{x:f,y:m};f=g.x,m=g.y;var _=r.hasOwnProperty("x"),b=r.hasOwnProperty("y"),v=Vt,y=zt,w=window;if(c){var A=$e(i),E="clientHeight",T="clientWidth";A===fe(i)&&"static"!==xe(A=Le(i)).position&&"absolute"===a&&(E="scrollHeight",T="scrollWidth"),(s===zt||(s===Vt||s===qt)&&o===Yt)&&(y=Rt,m-=(d&&A===w&&w.visualViewport?w.visualViewport.height:A[E])-n.height,m*=l?1:-1),s!==Vt&&(s!==zt&&s!==Rt||o!==Yt)||(v=qt,f-=(d&&A===w&&w.visualViewport?w.visualViewport.width:A[T])-n.width,f*=l?1:-1)}var C,O=Object.assign({position:a},c&&He),x=!0===h?function(t,e){var i=t.x,n=t.y,s=e.devicePixelRatio||1;return{x:we(i*s)/s||0,y:we(n*s)/s||0}}({x:f,y:m},fe(i)):{x:f,y:m};return f=x.x,m=x.y,l?Object.assign({},O,((C={})[y]=b?"0":"",C[v]=_?"0":"",C.transform=(w.devicePixelRatio||1)<=1?"translate("+f+"px, "+m+"px)":"translate3d("+f+"px, "+m+"px, 0)",C)):Object.assign({},O,((e={})[y]=b?m+"px":"",e[v]=_?f+"px":"",e.transform="",e))}const Be={name:"computeStyles",enabled:!0,phase:"beforeWrite",fn:function(t){var e=t.state,i=t.options,n=i.gpuAcceleration,s=void 0===n||n,o=i.adaptive,r=void 0===o||o,a=i.roundOffsets,l=void 0===a||a,c={placement:be(e.placement),variation:Fe(e.placement),popper:e.elements.popper,popperRect:e.rects.popper,gpuAcceleration:s,isFixed:"fixed"===e.options.strategy};null!=e.modifiersData.popperOffsets&&(e.styles.popper=Object.assign({},e.styles.popper,We(Object.assign({},c,{offsets:e.modifiersData.popperOffsets,position:e.options.strategy,adaptive:r,roundOffsets:l})))),null!=e.modifiersData.arrow&&(e.styles.arrow=Object.assign({},e.styles.arrow,We(Object.assign({},c,{offsets:e.modifiersData.arrow,position:"absolute",adaptive:!1,roundOffsets:l})))),e.attributes.popper=Object.assign({},e.attributes.popper,{"data-popper-placement":e.placement})},data:{}};var ze={passive:!0};const Re={name:"eventListeners",enabled:!0,phase:"write",fn:function(){},effect:function(t){var e=t.state,i=t.instance,n=t.options,s=n.scroll,o=void 0===s||s,r=n.resize,a=void 0===r||r,l=fe(e.elements.popper),c=[].concat(e.scrollParents.reference,e.scrollParents.popper);return o&&c.forEach((function(t){t.addEventListener("scroll",i.update,ze)})),a&&l.addEventListener("resize",i.update,ze),function(){o&&c.forEach((function(t){t.removeEventListener("scroll",i.update,ze)})),a&&l.removeEventListener("resize",i.update,ze)}},data:{}};var qe={left:"right",right:"left",bottom:"top",top:"bottom"};function Ve(t){return t.replace(/left|right|bottom|top/g,(function(t){return qe[t]}))}var Ke={start:"end",end:"start"};function Qe(t){return t.replace(/start|end/g,(function(t){return Ke[t]}))}function Xe(t){var e=fe(t);return{scrollLeft:e.pageXOffset,scrollTop:e.pageYOffset}}function Ye(t){return Te(Le(t)).left+Xe(t).scrollLeft}function Ue(t){var e=xe(t),i=e.overflow,n=e.overflowX,s=e.overflowY;return/auto|scroll|overlay|hidden/.test(i+s+n)}function Ge(t){return["html","body","#document"].indexOf(ue(t))>=0?t.ownerDocument.body:me(t)&&Ue(t)?t:Ge(Se(t))}function Je(t,e){var i;void 0===e&&(e=[]);var n=Ge(t),s=n===(null==(i=t.ownerDocument)?void 0:i.body),o=fe(n),r=s?[o].concat(o.visualViewport||[],Ue(n)?n:[]):n,a=e.concat(r);return s?a:a.concat(Je(Se(r)))}function Ze(t){return Object.assign({},t,{left:t.x,top:t.y,right:t.x+t.width,bottom:t.y+t.height})}function ti(t,e,i){return e===Gt?Ze(function(t,e){var i=fe(t),n=Le(t),s=i.visualViewport,o=n.clientWidth,r=n.clientHeight,a=0,l=0;if(s){o=s.width,r=s.height;var c=Ee();(c||!c&&"fixed"===e)&&(a=s.offsetLeft,l=s.offsetTop)}return{width:o,height:r,x:a+Ye(t),y:l}}(t,i)):pe(e)?function(t,e){var i=Te(t,!1,"fixed"===e);return i.top=i.top+t.clientTop,i.left=i.left+t.clientLeft,i.bottom=i.top+t.clientHeight,i.right=i.left+t.clientWidth,i.width=t.clientWidth,i.height=t.clientHeight,i.x=i.left,i.y=i.top,i}(e,i):Ze(function(t){var e,i=Le(t),n=Xe(t),s=null==(e=t.ownerDocument)?void 0:e.body,o=ve(i.scrollWidth,i.clientWidth,s?s.scrollWidth:0,s?s.clientWidth:0),r=ve(i.scrollHeight,i.clientHeight,s?s.scrollHeight:0,s?s.clientHeight:0),a=-n.scrollLeft+Ye(t),l=-n.scrollTop;return"rtl"===xe(s||i).direction&&(a+=ve(i.clientWidth,s?s.clientWidth:0)-o),{width:o,height:r,x:a,y:l}}(Le(t)))}function ei(t){var e,i=t.reference,n=t.element,s=t.placement,o=s?be(s):null,r=s?Fe(s):null,a=i.x+i.width/2-n.width/2,l=i.y+i.height/2-n.height/2;switch(o){case zt:e={x:a,y:i.y-n.height};break;case Rt:e={x:a,y:i.y+i.height};break;case qt:e={x:i.x+i.width,y:l};break;case Vt:e={x:i.x-n.width,y:l};break;default:e={x:i.x,y:i.y}}var c=o?Ie(o):null;if(null!=c){var h="y"===c?"height":"width";switch(r){case Xt:e[c]=e[c]-(i[h]/2-n[h]/2);break;case Yt:e[c]=e[c]+(i[h]/2-n[h]/2)}}return e}function ii(t,e){void 0===e&&(e={});var i=e,n=i.placement,s=void 0===n?t.placement:n,o=i.strategy,r=void 0===o?t.strategy:o,a=i.boundary,l=void 0===a?Ut:a,c=i.rootBoundary,h=void 0===c?Gt:c,d=i.elementContext,u=void 0===d?Jt:d,f=i.altBoundary,p=void 0!==f&&f,m=i.padding,g=void 0===m?0:m,_=Pe("number"!=typeof g?g:Me(g,Qt)),b=u===Jt?Zt:Jt,v=t.rects.popper,y=t.elements[p?b:u],w=function(t,e,i,n){var s="clippingParents"===e?function(t){var e=Je(Se(t)),i=["absolute","fixed"].indexOf(xe(t).position)>=0&&me(t)?$e(t):t;return pe(i)?e.filter((function(t){return pe(t)&&Oe(t,i)&&"body"!==ue(t)})):[]}(t):[].concat(e),o=[].concat(s,[i]),r=o[0],a=o.reduce((function(e,i){var s=ti(t,i,n);return e.top=ve(s.top,e.top),e.right=ye(s.right,e.right),e.bottom=ye(s.bottom,e.bottom),e.left=ve(s.left,e.left),e}),ti(t,r,n));return a.width=a.right-a.left,a.height=a.bottom-a.top,a.x=a.left,a.y=a.top,a}(pe(y)?y:y.contextElement||Le(t.elements.popper),l,h,r),A=Te(t.elements.reference),E=ei({reference:A,element:v,strategy:"absolute",placement:s}),T=Ze(Object.assign({},v,E)),C=u===Jt?T:A,O={top:w.top-C.top+_.top,bottom:C.bottom-w.bottom+_.bottom,left:w.left-C.left+_.left,right:C.right-w.right+_.right},x=t.modifiersData.offset;if(u===Jt&&x){var k=x[s];Object.keys(O).forEach((function(t){var e=[qt,Rt].indexOf(t)>=0?1:-1,i=[zt,Rt].indexOf(t)>=0?"y":"x";O[t]+=k[i]*e}))}return O}function ni(t,e){void 0===e&&(e={});var i=e,n=i.placement,s=i.boundary,o=i.rootBoundary,r=i.padding,a=i.flipVariations,l=i.allowedAutoPlacements,c=void 0===l?ee:l,h=Fe(n),d=h?a?te:te.filter((function(t){return Fe(t)===h})):Qt,u=d.filter((function(t){return c.indexOf(t)>=0}));0===u.length&&(u=d);var f=u.reduce((function(e,i){return e[i]=ii(t,{placement:i,boundary:s,rootBoundary:o,padding:r})[be(i)],e}),{});return Object.keys(f).sort((function(t,e){return f[t]-f[e]}))}const si={name:"flip",enabled:!0,phase:"main",fn:function(t){var e=t.state,i=t.options,n=t.name;if(!e.modifiersData[n]._skip){for(var s=i.mainAxis,o=void 0===s||s,r=i.altAxis,a=void 0===r||r,l=i.fallbackPlacements,c=i.padding,h=i.boundary,d=i.rootBoundary,u=i.altBoundary,f=i.flipVariations,p=void 0===f||f,m=i.allowedAutoPlacements,g=e.options.placement,_=be(g),b=l||(_!==g&&p?function(t){if(be(t)===Kt)return[];var e=Ve(t);return[Qe(t),e,Qe(e)]}(g):[Ve(g)]),v=[g].concat(b).reduce((function(t,i){return t.concat(be(i)===Kt?ni(e,{placement:i,boundary:h,rootBoundary:d,padding:c,flipVariations:p,allowedAutoPlacements:m}):i)}),[]),y=e.rects.reference,w=e.rects.popper,A=new Map,E=!0,T=v[0],C=0;C
+
+
+
+
+
+
+
+Decision Log
+ +
+
+
+
+ This document tracks major design decisions for the SCWorkflow package.
+
+
+2026-02-17: compareCellPopulations() Function
+Context: Need for visualizing cell population distributions across experimental groups (treatments, timepoints, conditions).
+Decision: Implemented dual visualization approach with alluvial flow plots and box plots.
+Rationale: - Alluvial flow plots show proportional relationships across groups while maintaining visibility of sample composition - Box plots provide statistical distribution view with individual data points - Both visualizations complement each other: flows for overall trends, boxes for statistical variance - Flexible frequency vs. counts mode supports different analytical needs
+Implementation Details: - Uses ggalluvial for flow visualizations (Sankey-style plots) - ggpubr for box plot styling with statistical comparisons - Internal helper createAnnoTable() aggregates frequencies/counts by sample - Supports custom color palettes or auto-generates from RColorBrewer - Handles group ordering for controlled presentation
Alternatives Considered: 1. Single bar plot visualization - rejected as insufficient for showing sample-level variation 2. Stacked area plots - rejected due to difficulty comparing non-adjacent categories 3. Separate samples in flow plots - rejected due to visual clutter with many samples
+Trade-offs: - Alluvial plots can be busy with many cell types (>10-15) - Box plots require sufficient samples per group for meaningful statistics - Two separate plots increase figure space requirements but provide complementary insights
+Generated From: JSON template Compare_Cell_Populations.code-template.json using json2r.prompt.md instructions
',title:"",trigger:"hover focus"},ls={allowList:"object",animation:"boolean",boundary:"(string|element)",container:"(string|element|boolean)",customClass:"(string|function)",delay:"(number|object)",fallbackPlacements:"array",html:"boolean",offset:"(array|string|function)",placement:"(string|function)",popperConfig:"(null|object|function)",sanitize:"boolean",sanitizeFn:"(null|function)",selector:"(string|boolean)",template:"string",title:"(string|element|function)",trigger:"string"};class cs extends W{constructor(t,e){if(void 0===vi)throw new TypeError("Bootstrap's tooltips require Popper (https://popper.js.org)");super(t,e),this._isEnabled=!0,this._timeout=0,this._isHovered=null,this._activeTrigger={},this._popper=null,this._templateFactory=null,this._newContent=null,this.tip=null,this._setListeners(),this._config.selector||this._fixTitle()}static get Default(){return as}static get DefaultType(){return ls}static get NAME(){return"tooltip"}enable(){this._isEnabled=!0}disable(){this._isEnabled=!1}toggleEnabled(){this._isEnabled=!this._isEnabled}toggle(){this._isEnabled&&(this._activeTrigger.click=!this._activeTrigger.click,this._isShown()?this._leave():this._enter())}dispose(){clearTimeout(this._timeout),N.off(this._element.closest(is),ns,this._hideModalHandler),this._element.getAttribute("data-bs-original-title")&&this._element.setAttribute("title",this._element.getAttribute("data-bs-original-title")),this._disposePopper(),super.dispose()}show(){if("none"===this._element.style.display)throw new Error("Please use show on visible elements");if(!this._isWithContent()||!this._isEnabled)return;const t=N.trigger(this._element,this.constructor.eventName("show")),e=(c(this._element)||this._element.ownerDocument.documentElement).contains(this._element);if(t.defaultPrevented||!e)return;this._disposePopper();const i=this._getTipElement();this._element.setAttribute("aria-describedby",i.getAttribute("id"));const{container:n}=this._config;if(this._element.ownerDocument.documentElement.contains(this.tip)||(n.append(i),N.trigger(this._element,this.constructor.eventName("inserted"))),this._popper=this._createPopper(i),i.classList.add(es),"ontouchstart"in document.documentElement)for(const t of[].concat(...document.body.children))N.on(t,"mouseover",h);this._queueCallback((()=>{N.trigger(this._element,this.constructor.eventName("shown")),!1===this._isHovered&&this._leave(),this._isHovered=!1}),this.tip,this._isAnimated())}hide(){if(this._isShown()&&!N.trigger(this._element,this.constructor.eventName("hide")).defaultPrevented){if(this._getTipElement().classList.remove(es),"ontouchstart"in document.documentElement)for(const t of[].concat(...document.body.children))N.off(t,"mouseover",h);this._activeTrigger.click=!1,this._activeTrigger[os]=!1,this._activeTrigger[ss]=!1,this._isHovered=null,this._queueCallback((()=>{this._isWithActiveTrigger()||(this._isHovered||this._disposePopper(),this._element.removeAttribute("aria-describedby"),N.trigger(this._element,this.constructor.eventName("hidden")))}),this.tip,this._isAnimated())}}update(){this._popper&&this._popper.update()}_isWithContent(){return Boolean(this._getTitle())}_getTipElement(){return this.tip||(this.tip=this._createTipElement(this._newContent||this._getContentForTemplate())),this.tip}_createTipElement(t){const e=this._getTemplateFactory(t).toHtml();if(!e)return null;e.classList.remove(ts,es),e.classList.add(`bs-${this.constructor.NAME}-auto`);const i=(t=>{do{t+=Math.floor(1e6*Math.random())}while(document.getElementById(t));return t})(this.constructor.NAME).toString();return e.setAttribute("id",i),this._isAnimated()&&e.classList.add(ts),e}setContent(t){this._newContent=t,this._isShown()&&(this._disposePopper(),this.show())}_getTemplateFactory(t){return this._templateFactory?this._templateFactory.changeContent(t):this._templateFactory=new Jn({...this._config,content:t,extraClass:this._resolvePossibleFunction(this._config.customClass)}),this._templateFactory}_getContentForTemplate(){return{".tooltip-inner":this._getTitle()}}_getTitle(){return this._resolvePossibleFunction(this._config.title)||this._element.getAttribute("data-bs-original-title")}_initializeOnDelegatedTarget(t){return this.constructor.getOrCreateInstance(t.delegateTarget,this._getDelegateConfig())}_isAnimated(){return this._config.animation||this.tip&&this.tip.classList.contains(ts)}_isShown(){return this.tip&&this.tip.classList.contains(es)}_createPopper(t){const e=g(this._config.placement,[this,t,this._element]),i=rs[e.toUpperCase()];return bi(this._element,t,this._getPopperConfig(i))}_getOffset(){const{offset:t}=this._config;return"string"==typeof t?t.split(",").map((t=>Number.parseInt(t,10))):"function"==typeof t?e=>t(e,this._element):t}_resolvePossibleFunction(t){return g(t,[this._element])}_getPopperConfig(t){const e={placement:t,modifiers:[{name:"flip",options:{fallbackPlacements:this._config.fallbackPlacements}},{name:"offset",options:{offset:this._getOffset()}},{name:"preventOverflow",options:{boundary:this._config.boundary}},{name:"arrow",options:{element:`.${this.constructor.NAME}-arrow`}},{name:"preSetPlacement",enabled:!0,phase:"beforeMain",fn:t=>{this._getTipElement().setAttribute("data-popper-placement",t.state.placement)}}]};return{...e,...g(this._config.popperConfig,[e])}}_setListeners(){const t=this._config.trigger.split(" ");for(const e of t)if("click"===e)N.on(this._element,this.constructor.eventName("click"),this._config.selector,(t=>{this._initializeOnDelegatedTarget(t).toggle()}));else if("manual"!==e){const t=e===ss?this.constructor.eventName("mouseenter"):this.constructor.eventName("focusin"),i=e===ss?this.constructor.eventName("mouseleave"):this.constructor.eventName("focusout");N.on(this._element,t,this._config.selector,(t=>{const e=this._initializeOnDelegatedTarget(t);e._activeTrigger["focusin"===t.type?os:ss]=!0,e._enter()})),N.on(this._element,i,this._config.selector,(t=>{const e=this._initializeOnDelegatedTarget(t);e._activeTrigger["focusout"===t.type?os:ss]=e._element.contains(t.relatedTarget),e._leave()}))}this._hideModalHandler=()=>{this._element&&this.hide()},N.on(this._element.closest(is),ns,this._hideModalHandler)}_fixTitle(){const t=this._element.getAttribute("title");t&&(this._element.getAttribute("aria-label")||this._element.textContent.trim()||this._element.setAttribute("aria-label",t),this._element.setAttribute("data-bs-original-title",t),this._element.removeAttribute("title"))}_enter(){this._isShown()||this._isHovered?this._isHovered=!0:(this._isHovered=!0,this._setTimeout((()=>{this._isHovered&&this.show()}),this._config.delay.show))}_leave(){this._isWithActiveTrigger()||(this._isHovered=!1,this._setTimeout((()=>{this._isHovered||this.hide()}),this._config.delay.hide))}_setTimeout(t,e){clearTimeout(this._timeout),this._timeout=setTimeout(t,e)}_isWithActiveTrigger(){return Object.values(this._activeTrigger).includes(!0)}_getConfig(t){const e=F.getDataAttributes(this._element);for(const t of Object.keys(e))Zn.has(t)&&delete e[t];return t={...e,..."object"==typeof t&&t?t:{}},t=this._mergeConfigObj(t),t=this._configAfterMerge(t),this._typeCheckConfig(t),t}_configAfterMerge(t){return t.container=!1===t.container?document.body:r(t.container),"number"==typeof t.delay&&(t.delay={show:t.delay,hide:t.delay}),"number"==typeof t.title&&(t.title=t.title.toString()),"number"==typeof t.content&&(t.content=t.content.toString()),t}_getDelegateConfig(){const t={};for(const[e,i]of Object.entries(this._config))this.constructor.Default[e]!==i&&(t[e]=i);return t.selector=!1,t.trigger="manual",t}_disposePopper(){this._popper&&(this._popper.destroy(),this._popper=null),this.tip&&(this.tip.remove(),this.tip=null)}static jQueryInterface(t){return this.each((function(){const e=cs.getOrCreateInstance(this,t);if("string"==typeof t){if(void 0===e[t])throw new TypeError(`No method named "${t}"`);e[t]()}}))}}m(cs);const hs={...cs.Default,content:"",offset:[0,8],placement:"right",template:'
',trigger:"click"},ds={...cs.DefaultType,content:"(null|string|element|function)"};class us extends cs{static get Default(){return hs}static get DefaultType(){return ds}static get NAME(){return"popover"}_isWithContent(){return this._getTitle()||this._getContent()}_getContentForTemplate(){return{".popover-header":this._getTitle(),".popover-body":this._getContent()}}_getContent(){return this._resolvePossibleFunction(this._config.content)}static jQueryInterface(t){return this.each((function(){const e=us.getOrCreateInstance(this,t);if("string"==typeof t){if(void 0===e[t])throw new TypeError(`No method named "${t}"`);e[t]()}}))}}m(us);const fs=".bs.scrollspy",ps=`activate${fs}`,ms=`click${fs}`,gs=`load${fs}.data-api`,_s="active",bs="[href]",vs=".nav-link",ys=`${vs}, .nav-item > ${vs}, .list-group-item`,ws={offset:null,rootMargin:"0px 0px -25%",smoothScroll:!1,target:null,threshold:[.1,.5,1]},As={offset:"(number|null)",rootMargin:"string",smoothScroll:"boolean",target:"element",threshold:"array"};class Es extends W{constructor(t,e){super(t,e),this._targetLinks=new Map,this._observableSections=new Map,this._rootElement="visible"===getComputedStyle(this._element).overflowY?null:this._element,this._activeTarget=null,this._observer=null,this._previousScrollData={visibleEntryTop:0,parentScrollTop:0},this.refresh()}static get Default(){return ws}static get DefaultType(){return As}static get NAME(){return"scrollspy"}refresh(){this._initializeTargetsAndObservables(),this._maybeEnableSmoothScroll(),this._observer?this._observer.disconnect():this._observer=this._getNewObserver();for(const t of this._observableSections.values())this._observer.observe(t)}dispose(){this._observer.disconnect(),super.dispose()}_configAfterMerge(t){return t.target=r(t.target)||document.body,t.rootMargin=t.offset?`${t.offset}px 0px -30%`:t.rootMargin,"string"==typeof t.threshold&&(t.threshold=t.threshold.split(",").map((t=>Number.parseFloat(t)))),t}_maybeEnableSmoothScroll(){this._config.smoothScroll&&(N.off(this._config.target,ms),N.on(this._config.target,ms,bs,(t=>{const e=this._observableSections.get(t.target.hash);if(e){t.preventDefault();const i=this._rootElement||window,n=e.offsetTop-this._element.offsetTop;if(i.scrollTo)return void i.scrollTo({top:n,behavior:"smooth"});i.scrollTop=n}})))}_getNewObserver(){const t={root:this._rootElement,threshold:this._config.threshold,rootMargin:this._config.rootMargin};return new IntersectionObserver((t=>this._observerCallback(t)),t)}_observerCallback(t){const e=t=>this._targetLinks.get(`#${t.target.id}`),i=t=>{this._previousScrollData.visibleEntryTop=t.target.offsetTop,this._process(e(t))},n=(this._rootElement||document.documentElement).scrollTop,s=n>=this._previousScrollData.parentScrollTop;this._previousScrollData.parentScrollTop=n;for(const o of t){if(!o.isIntersecting){this._activeTarget=null,this._clearActiveClass(e(o));continue}const t=o.target.offsetTop>=this._previousScrollData.visibleEntryTop;if(s&&t){if(i(o),!n)return}else s||t||i(o)}}_initializeTargetsAndObservables(){this._targetLinks=new Map,this._observableSections=new Map;const t=z.find(bs,this._config.target);for(const e of t){if(!e.hash||l(e))continue;const t=z.findOne(decodeURI(e.hash),this._element);a(t)&&(this._targetLinks.set(decodeURI(e.hash),e),this._observableSections.set(e.hash,t))}}_process(t){this._activeTarget!==t&&(this._clearActiveClass(this._config.target),this._activeTarget=t,t.classList.add(_s),this._activateParents(t),N.trigger(this._element,ps,{relatedTarget:t}))}_activateParents(t){if(t.classList.contains("dropdown-item"))z.findOne(".dropdown-toggle",t.closest(".dropdown")).classList.add(_s);else for(const e of z.parents(t,".nav, .list-group"))for(const t of z.prev(e,ys))t.classList.add(_s)}_clearActiveClass(t){t.classList.remove(_s);const e=z.find(`${bs}.${_s}`,t);for(const t of e)t.classList.remove(_s)}static jQueryInterface(t){return this.each((function(){const e=Es.getOrCreateInstance(this,t);if("string"==typeof t){if(void 0===e[t]||t.startsWith("_")||"constructor"===t)throw new TypeError(`No method named "${t}"`);e[t]()}}))}}N.on(window,gs,(()=>{for(const t of z.find('[data-bs-spy="scroll"]'))Es.getOrCreateInstance(t)})),m(Es);const Ts=".bs.tab",Cs=`hide${Ts}`,Os=`hidden${Ts}`,xs=`show${Ts}`,ks=`shown${Ts}`,Ls=`click${Ts}`,Ss=`keydown${Ts}`,Ds=`load${Ts}`,$s="ArrowLeft",Is="ArrowRight",Ns="ArrowUp",Ps="ArrowDown",Ms="Home",js="End",Fs="active",Hs="fade",Ws="show",Bs=":not(.dropdown-toggle)",zs='[data-bs-toggle="tab"], [data-bs-toggle="pill"], [data-bs-toggle="list"]',Rs=`.nav-link${Bs}, .list-group-item${Bs}, [role="tab"]${Bs}, ${zs}`,qs=`.${Fs}[data-bs-toggle="tab"], .${Fs}[data-bs-toggle="pill"], .${Fs}[data-bs-toggle="list"]`;class Vs extends W{constructor(t){super(t),this._parent=this._element.closest('.list-group, .nav, [role="tablist"]'),this._parent&&(this._setInitialAttributes(this._parent,this._getChildren()),N.on(this._element,Ss,(t=>this._keydown(t))))}static get NAME(){return"tab"}show(){const t=this._element;if(this._elemIsActive(t))return;const e=this._getActiveElem(),i=e?N.trigger(e,Cs,{relatedTarget:t}):null;N.trigger(t,xs,{relatedTarget:e}).defaultPrevented||i&&i.defaultPrevented||(this._deactivate(e,t),this._activate(t,e))}_activate(t,e){t&&(t.classList.add(Fs),this._activate(z.getElementFromSelector(t)),this._queueCallback((()=>{"tab"===t.getAttribute("role")?(t.removeAttribute("tabindex"),t.setAttribute("aria-selected",!0),this._toggleDropDown(t,!0),N.trigger(t,ks,{relatedTarget:e})):t.classList.add(Ws)}),t,t.classList.contains(Hs)))}_deactivate(t,e){t&&(t.classList.remove(Fs),t.blur(),this._deactivate(z.getElementFromSelector(t)),this._queueCallback((()=>{"tab"===t.getAttribute("role")?(t.setAttribute("aria-selected",!1),t.setAttribute("tabindex","-1"),this._toggleDropDown(t,!1),N.trigger(t,Os,{relatedTarget:e})):t.classList.remove(Ws)}),t,t.classList.contains(Hs)))}_keydown(t){if(![$s,Is,Ns,Ps,Ms,js].includes(t.key))return;t.stopPropagation(),t.preventDefault();const e=this._getChildren().filter((t=>!l(t)));let i;if([Ms,js].includes(t.key))i=e[t.key===Ms?0:e.length-1];else{const n=[Is,Ps].includes(t.key);i=b(e,t.target,n,!0)}i&&(i.focus({preventScroll:!0}),Vs.getOrCreateInstance(i).show())}_getChildren(){return z.find(Rs,this._parent)}_getActiveElem(){return this._getChildren().find((t=>this._elemIsActive(t)))||null}_setInitialAttributes(t,e){this._setAttributeIfNotExists(t,"role","tablist");for(const t of e)this._setInitialAttributesOnChild(t)}_setInitialAttributesOnChild(t){t=this._getInnerElement(t);const e=this._elemIsActive(t),i=this._getOuterElement(t);t.setAttribute("aria-selected",e),i!==t&&this._setAttributeIfNotExists(i,"role","presentation"),e||t.setAttribute("tabindex","-1"),this._setAttributeIfNotExists(t,"role","tab"),this._setInitialAttributesOnTargetPanel(t)}_setInitialAttributesOnTargetPanel(t){const e=z.getElementFromSelector(t);e&&(this._setAttributeIfNotExists(e,"role","tabpanel"),t.id&&this._setAttributeIfNotExists(e,"aria-labelledby",`${t.id}`))}_toggleDropDown(t,e){const i=this._getOuterElement(t);if(!i.classList.contains("dropdown"))return;const n=(t,n)=>{const s=z.findOne(t,i);s&&s.classList.toggle(n,e)};n(".dropdown-toggle",Fs),n(".dropdown-menu",Ws),i.setAttribute("aria-expanded",e)}_setAttributeIfNotExists(t,e,i){t.hasAttribute(e)||t.setAttribute(e,i)}_elemIsActive(t){return t.classList.contains(Fs)}_getInnerElement(t){return t.matches(Rs)?t:z.findOne(Rs,t)}_getOuterElement(t){return t.closest(".nav-item, .list-group-item")||t}static jQueryInterface(t){return this.each((function(){const e=Vs.getOrCreateInstance(this);if("string"==typeof t){if(void 0===e[t]||t.startsWith("_")||"constructor"===t)throw new TypeError(`No method named "${t}"`);e[t]()}}))}}N.on(document,Ls,zs,(function(t){["A","AREA"].includes(this.tagName)&&t.preventDefault(),l(this)||Vs.getOrCreateInstance(this).show()})),N.on(window,Ds,(()=>{for(const t of z.find(qs))Vs.getOrCreateInstance(t)})),m(Vs);const Ks=".bs.toast",Qs=`mouseover${Ks}`,Xs=`mouseout${Ks}`,Ys=`focusin${Ks}`,Us=`focusout${Ks}`,Gs=`hide${Ks}`,Js=`hidden${Ks}`,Zs=`show${Ks}`,to=`shown${Ks}`,eo="hide",io="show",no="showing",so={animation:"boolean",autohide:"boolean",delay:"number"},oo={animation:!0,autohide:!0,delay:5e3};class ro extends W{constructor(t,e){super(t,e),this._timeout=null,this._hasMouseInteraction=!1,this._hasKeyboardInteraction=!1,this._setListeners()}static get Default(){return oo}static get DefaultType(){return so}static get NAME(){return"toast"}show(){N.trigger(this._element,Zs).defaultPrevented||(this._clearTimeout(),this._config.animation&&this._element.classList.add("fade"),this._element.classList.remove(eo),d(this._element),this._element.classList.add(io,no),this._queueCallback((()=>{this._element.classList.remove(no),N.trigger(this._element,to),this._maybeScheduleHide()}),this._element,this._config.animation))}hide(){this.isShown()&&(N.trigger(this._element,Gs).defaultPrevented||(this._element.classList.add(no),this._queueCallback((()=>{this._element.classList.add(eo),this._element.classList.remove(no,io),N.trigger(this._element,Js)}),this._element,this._config.animation)))}dispose(){this._clearTimeout(),this.isShown()&&this._element.classList.remove(io),super.dispose()}isShown(){return this._element.classList.contains(io)}_maybeScheduleHide(){this._config.autohide&&(this._hasMouseInteraction||this._hasKeyboardInteraction||(this._timeout=setTimeout((()=>{this.hide()}),this._config.delay)))}_onInteraction(t,e){switch(t.type){case"mouseover":case"mouseout":this._hasMouseInteraction=e;break;case"focusin":case"focusout":this._hasKeyboardInteraction=e}if(e)return void this._clearTimeout();const i=t.relatedTarget;this._element===i||this._element.contains(i)||this._maybeScheduleHide()}_setListeners(){N.on(this._element,Qs,(t=>this._onInteraction(t,!0))),N.on(this._element,Xs,(t=>this._onInteraction(t,!1))),N.on(this._element,Ys,(t=>this._onInteraction(t,!0))),N.on(this._element,Us,(t=>this._onInteraction(t,!1)))}_clearTimeout(){clearTimeout(this._timeout),this._timeout=null}static jQueryInterface(t){return this.each((function(){const e=ro.getOrCreateInstance(this,t);if("string"==typeof t){if(void 0===e[t])throw new TypeError(`No method named "${t}"`);e[t](this)}}))}}return R(ro),m(ro),{Alert:Q,Button:Y,Carousel:xt,Collapse:Bt,Dropdown:qi,Modal:On,Offcanvas:qn,Popover:us,ScrollSpy:Es,Tab:Vs,Toast:ro,Tooltip:cs}}));
+//# sourceMappingURL=bootstrap.bundle.min.js.map
\ No newline at end of file
diff --git a/docs/deps/bootstrap-5.3.1/bootstrap.bundle.min.js.map b/docs/deps/bootstrap-5.3.1/bootstrap.bundle.min.js.map
new file mode 100644
index 0000000..3863da8
--- /dev/null
+++ b/docs/deps/bootstrap-5.3.1/bootstrap.bundle.min.js.map
@@ -0,0 +1 @@
+{"version":3,"names":["elementMap","Map","Data","set","element","key","instance","has","instanceMap","get","size","console","error","Array","from","keys","remove","delete","TRANSITION_END","parseSelector","selector","window","CSS","escape","replace","match","id","triggerTransitionEnd","dispatchEvent","Event","isElement","object","jquery","nodeType","getElement","length","document","querySelector","isVisible","getClientRects","elementIsVisible","getComputedStyle","getPropertyValue","closedDetails","closest","summary","parentNode","isDisabled","Node","ELEMENT_NODE","classList","contains","disabled","hasAttribute","getAttribute","findShadowRoot","documentElement","attachShadow","getRootNode","root","ShadowRoot","noop","reflow","offsetHeight","getjQuery","jQuery","body","DOMContentLoadedCallbacks","isRTL","dir","defineJQueryPlugin","plugin","callback","$","name","NAME","JQUERY_NO_CONFLICT","fn","jQueryInterface","Constructor","noConflict","readyState","addEventListener","push","execute","possibleCallback","args","defaultValue","executeAfterTransition","transitionElement","waitForTransition","emulatedDuration","transitionDuration","transitionDelay","floatTransitionDuration","Number","parseFloat","floatTransitionDelay","split","getTransitionDurationFromElement","called","handler","target","removeEventListener","setTimeout","getNextActiveElement","list","activeElement","shouldGetNext","isCycleAllowed","listLength","index","indexOf","Math","max","min","namespaceRegex","stripNameRegex","stripUidRegex","eventRegistry","uidEvent","customEvents","mouseenter","mouseleave","nativeEvents","Set","makeEventUid","uid","getElementEvents","findHandler","events","callable","delegationSelector","Object","values","find","event","normalizeParameters","originalTypeEvent","delegationFunction","isDelegated","typeEvent","getTypeEvent","addHandler","oneOff","wrapFunction","relatedTarget","delegateTarget","call","this","handlers","previousFunction","domElements","querySelectorAll","domElement","hydrateObj","EventHandler","off","type","apply","bootstrapDelegationHandler","bootstrapHandler","removeHandler","Boolean","removeNamespacedHandlers","namespace","storeElementEvent","handlerKey","entries","includes","on","one","inNamespace","isNamespace","startsWith","elementEvent","slice","keyHandlers","trigger","jQueryEvent","bubbles","nativeDispatch","defaultPrevented","isPropagationStopped","isImmediatePropagationStopped","isDefaultPrevented","evt","cancelable","preventDefault","obj","meta","value","_unused","defineProperty","configurable","normalizeData","toString","JSON","parse","decodeURIComponent","normalizeDataKey","chr","toLowerCase","Manipulator","setDataAttribute","setAttribute","removeDataAttribute","removeAttribute","getDataAttributes","attributes","bsKeys","dataset","filter","pureKey","charAt","getDataAttribute","Config","Default","DefaultType","Error","_getConfig","config","_mergeConfigObj","_configAfterMerge","_typeCheckConfig","jsonConfig","constructor","configTypes","property","expectedTypes","valueType","prototype","RegExp","test","TypeError","toUpperCase","BaseComponent","super","_element","_config","DATA_KEY","dispose","EVENT_KEY","propertyName","getOwnPropertyNames","_queueCallback","isAnimated","getInstance","getOrCreateInstance","VERSION","eventName","getSelector","hrefAttribute","trim","SelectorEngine","concat","Element","findOne","children","child","matches","parents","ancestor","prev","previous","previousElementSibling","next","nextElementSibling","focusableChildren","focusables","map","join","el","getSelectorFromElement","getElementFromSelector","getMultipleElementsFromSelector","enableDismissTrigger","component","method","clickEvent","tagName","EVENT_CLOSE","EVENT_CLOSED","Alert","close","_destroyElement","each","data","undefined","SELECTOR_DATA_TOGGLE","Button","toggle","button","EVENT_TOUCHSTART","EVENT_TOUCHMOVE","EVENT_TOUCHEND","EVENT_POINTERDOWN","EVENT_POINTERUP","endCallback","leftCallback","rightCallback","Swipe","isSupported","_deltaX","_supportPointerEvents","PointerEvent","_initEvents","_start","_eventIsPointerPenTouch","clientX","touches","_end","_handleSwipe","_move","absDeltaX","abs","direction","add","pointerType","navigator","maxTouchPoints","DATA_API_KEY","ORDER_NEXT","ORDER_PREV","DIRECTION_LEFT","DIRECTION_RIGHT","EVENT_SLIDE","EVENT_SLID","EVENT_KEYDOWN","EVENT_MOUSEENTER","EVENT_MOUSELEAVE","EVENT_DRAG_START","EVENT_LOAD_DATA_API","EVENT_CLICK_DATA_API","CLASS_NAME_CAROUSEL","CLASS_NAME_ACTIVE","SELECTOR_ACTIVE","SELECTOR_ITEM","SELECTOR_ACTIVE_ITEM","KEY_TO_DIRECTION","ArrowLeft","ArrowRight","interval","keyboard","pause","ride","touch","wrap","Carousel","_interval","_activeElement","_isSliding","touchTimeout","_swipeHelper","_indicatorsElement","_addEventListeners","cycle","_slide","nextWhenVisible","hidden","_clearInterval","_updateInterval","setInterval","_maybeEnableCycle","to","items","_getItems","activeIndex","_getItemIndex","_getActive","order","defaultInterval","_keydown","_addTouchEventListeners","img","swipeConfig","_directionToOrder","endCallBack","clearTimeout","_setActiveIndicatorElement","activeIndicator","newActiveIndicator","elementInterval","parseInt","isNext","nextElement","nextElementIndex","triggerEvent","_orderToDirection","isCycling","directionalClassName","orderClassName","completeCallBack","_isAnimated","clearInterval","carousel","slideIndex","carousels","EVENT_SHOW","EVENT_SHOWN","EVENT_HIDE","EVENT_HIDDEN","CLASS_NAME_SHOW","CLASS_NAME_COLLAPSE","CLASS_NAME_COLLAPSING","CLASS_NAME_DEEPER_CHILDREN","parent","Collapse","_isTransitioning","_triggerArray","toggleList","elem","filterElement","foundElement","_initializeChildren","_addAriaAndCollapsedClass","_isShown","hide","show","activeChildren","_getFirstLevelChildren","activeInstance","dimension","_getDimension","style","scrollSize","complete","getBoundingClientRect","selected","triggerArray","isOpen","top","bottom","right","left","auto","basePlacements","start","end","clippingParents","viewport","popper","reference","variationPlacements","reduce","acc","placement","placements","beforeRead","read","afterRead","beforeMain","main","afterMain","beforeWrite","write","afterWrite","modifierPhases","getNodeName","nodeName","getWindow","node","ownerDocument","defaultView","isHTMLElement","HTMLElement","isShadowRoot","applyStyles$1","enabled","phase","_ref","state","elements","forEach","styles","assign","effect","_ref2","initialStyles","position","options","strategy","margin","arrow","hasOwnProperty","attribute","requires","getBasePlacement","round","getUAString","uaData","userAgentData","brands","isArray","item","brand","version","userAgent","isLayoutViewport","includeScale","isFixedStrategy","clientRect","scaleX","scaleY","offsetWidth","width","height","visualViewport","addVisualOffsets","x","offsetLeft","y","offsetTop","getLayoutRect","rootNode","isSameNode","host","isTableElement","getDocumentElement","getParentNode","assignedSlot","getTrueOffsetParent","offsetParent","getOffsetParent","isFirefox","currentNode","css","transform","perspective","contain","willChange","getContainingBlock","getMainAxisFromPlacement","within","mathMax","mathMin","mergePaddingObject","paddingObject","expandToHashMap","hashMap","arrow$1","_state$modifiersData$","arrowElement","popperOffsets","modifiersData","basePlacement","axis","len","padding","rects","toPaddingObject","arrowRect","minProp","maxProp","endDiff","startDiff","arrowOffsetParent","clientSize","clientHeight","clientWidth","centerToReference","center","offset","axisProp","centerOffset","_options$element","requiresIfExists","getVariation","unsetSides","mapToStyles","_Object$assign2","popperRect","variation","offsets","gpuAcceleration","adaptive","roundOffsets","isFixed","_offsets$x","_offsets$y","_ref3","hasX","hasY","sideX","sideY","win","heightProp","widthProp","_Object$assign","commonStyles","_ref4","dpr","devicePixelRatio","roundOffsetsByDPR","computeStyles$1","_ref5","_options$gpuAccelerat","_options$adaptive","_options$roundOffsets","passive","eventListeners","_options$scroll","scroll","_options$resize","resize","scrollParents","scrollParent","update","hash","getOppositePlacement","matched","getOppositeVariationPlacement","getWindowScroll","scrollLeft","pageXOffset","scrollTop","pageYOffset","getWindowScrollBarX","isScrollParent","_getComputedStyle","overflow","overflowX","overflowY","getScrollParent","listScrollParents","_element$ownerDocumen","isBody","updatedList","rectToClientRect","rect","getClientRectFromMixedType","clippingParent","html","layoutViewport","getViewportRect","clientTop","clientLeft","getInnerBoundingClientRect","winScroll","scrollWidth","scrollHeight","getDocumentRect","computeOffsets","commonX","commonY","mainAxis","detectOverflow","_options","_options$placement","_options$strategy","_options$boundary","boundary","_options$rootBoundary","rootBoundary","_options$elementConte","elementContext","_options$altBoundary","altBoundary","_options$padding","altContext","clippingClientRect","mainClippingParents","clipperElement","getClippingParents","firstClippingParent","clippingRect","accRect","getClippingRect","contextElement","referenceClientRect","popperClientRect","elementClientRect","overflowOffsets","offsetData","multiply","computeAutoPlacement","flipVariations","_options$allowedAutoP","allowedAutoPlacements","allPlacements","allowedPlacements","overflows","sort","a","b","flip$1","_skip","_options$mainAxis","checkMainAxis","_options$altAxis","altAxis","checkAltAxis","specifiedFallbackPlacements","fallbackPlacements","_options$flipVariatio","preferredPlacement","oppositePlacement","getExpandedFallbackPlacements","referenceRect","checksMap","makeFallbackChecks","firstFittingPlacement","i","_basePlacement","isStartVariation","isVertical","mainVariationSide","altVariationSide","checks","every","check","_loop","_i","fittingPlacement","reset","getSideOffsets","preventedOffsets","isAnySideFullyClipped","some","side","hide$1","preventOverflow","referenceOverflow","popperAltOverflow","referenceClippingOffsets","popperEscapeOffsets","isReferenceHidden","hasPopperEscaped","offset$1","_options$offset","invertDistance","skidding","distance","distanceAndSkiddingToXY","_data$state$placement","popperOffsets$1","preventOverflow$1","_options$tether","tether","_options$tetherOffset","tetherOffset","isBasePlacement","tetherOffsetValue","normalizedTetherOffsetValue","offsetModifierState","_offsetModifierState$","mainSide","altSide","additive","minLen","maxLen","arrowPaddingObject","arrowPaddingMin","arrowPaddingMax","arrowLen","minOffset","maxOffset","clientOffset","offsetModifierValue","tetherMax","preventedOffset","_offsetModifierState$2","_mainSide","_altSide","_offset","_len","_min","_max","isOriginSide","_offsetModifierValue","_tetherMin","_tetherMax","_preventedOffset","v","withinMaxClamp","getCompositeRect","elementOrVirtualElement","isOffsetParentAnElement","offsetParentIsScaled","isElementScaled","modifiers","visited","result","modifier","dep","depModifier","DEFAULT_OPTIONS","areValidElements","arguments","_key","popperGenerator","generatorOptions","_generatorOptions","_generatorOptions$def","defaultModifiers","_generatorOptions$def2","defaultOptions","pending","orderedModifiers","effectCleanupFns","isDestroyed","setOptions","setOptionsAction","cleanupModifierEffects","merged","orderModifiers","current","existing","m","_ref$options","cleanupFn","forceUpdate","_state$elements","_state$orderedModifie","_state$orderedModifie2","Promise","resolve","then","destroy","onFirstUpdate","createPopper","computeStyles","applyStyles","flip","ARROW_UP_KEY","ARROW_DOWN_KEY","EVENT_KEYDOWN_DATA_API","EVENT_KEYUP_DATA_API","SELECTOR_DATA_TOGGLE_SHOWN","SELECTOR_MENU","PLACEMENT_TOP","PLACEMENT_TOPEND","PLACEMENT_BOTTOM","PLACEMENT_BOTTOMEND","PLACEMENT_RIGHT","PLACEMENT_LEFT","autoClose","display","popperConfig","Dropdown","_popper","_parent","_menu","_inNavbar","_detectNavbar","_createPopper","focus","_completeHide","Popper","referenceElement","_getPopperConfig","_getPlacement","parentDropdown","isEnd","_getOffset","popperData","defaultBsPopperConfig","_selectMenuItem","clearMenus","openToggles","context","composedPath","isMenuTarget","dataApiKeydownHandler","isInput","isEscapeEvent","isUpOrDownEvent","getToggleButton","stopPropagation","EVENT_MOUSEDOWN","className","clickCallback","rootElement","Backdrop","_isAppended","_append","_getElement","_emulateAnimation","backdrop","createElement","append","EVENT_FOCUSIN","EVENT_KEYDOWN_TAB","TAB_NAV_BACKWARD","autofocus","trapElement","FocusTrap","_isActive","_lastTabNavDirection","activate","_handleFocusin","_handleKeydown","deactivate","shiftKey","SELECTOR_FIXED_CONTENT","SELECTOR_STICKY_CONTENT","PROPERTY_PADDING","PROPERTY_MARGIN","ScrollBarHelper","getWidth","documentWidth","innerWidth","_disableOverFlow","_setElementAttributes","calculatedValue","_resetElementAttributes","isOverflowing","_saveInitialAttribute","styleProperty","scrollbarWidth","_applyManipulationCallback","setProperty","actualValue","removeProperty","callBack","sel","EVENT_HIDE_PREVENTED","EVENT_RESIZE","EVENT_CLICK_DISMISS","EVENT_MOUSEDOWN_DISMISS","EVENT_KEYDOWN_DISMISS","CLASS_NAME_OPEN","CLASS_NAME_STATIC","Modal","_dialog","_backdrop","_initializeBackDrop","_focustrap","_initializeFocusTrap","_scrollBar","_adjustDialog","_showElement","_hideModal","handleUpdate","modalBody","transitionComplete","_triggerBackdropTransition","event2","_resetAdjustments","isModalOverflowing","initialOverflowY","isBodyOverflowing","paddingLeft","paddingRight","showEvent","alreadyOpen","CLASS_NAME_SHOWING","CLASS_NAME_HIDING","OPEN_SELECTOR","Offcanvas","blur","completeCallback","DefaultAllowlist","area","br","col","code","div","em","hr","h1","h2","h3","h4","h5","h6","li","ol","p","pre","s","small","span","sub","sup","strong","u","ul","uriAttributes","SAFE_URL_PATTERN","allowedAttribute","allowedAttributeList","attributeName","nodeValue","attributeRegex","regex","allowList","content","extraClass","sanitize","sanitizeFn","template","DefaultContentType","entry","TemplateFactory","getContent","_resolvePossibleFunction","hasContent","changeContent","_checkContent","toHtml","templateWrapper","innerHTML","_maybeSanitize","text","_setContent","arg","templateElement","_putElementInTemplate","textContent","unsafeHtml","sanitizeFunction","createdDocument","DOMParser","parseFromString","elementName","attributeList","allowedAttributes","sanitizeHtml","DISALLOWED_ATTRIBUTES","CLASS_NAME_FADE","SELECTOR_MODAL","EVENT_MODAL_HIDE","TRIGGER_HOVER","TRIGGER_FOCUS","AttachmentMap","AUTO","TOP","RIGHT","BOTTOM","LEFT","animation","container","customClass","delay","title","Tooltip","_isEnabled","_timeout","_isHovered","_activeTrigger","_templateFactory","_newContent","tip","_setListeners","_fixTitle","enable","disable","toggleEnabled","click","_leave","_enter","_hideModalHandler","_disposePopper","_isWithContent","isInTheDom","_getTipElement","_isWithActiveTrigger","_getTitle","_createTipElement","_getContentForTemplate","_getTemplateFactory","tipId","prefix","floor","random","getElementById","getUID","setContent","_initializeOnDelegatedTarget","_getDelegateConfig","attachment","triggers","eventIn","eventOut","_setTimeout","timeout","dataAttributes","dataAttribute","Popover","_getContent","EVENT_ACTIVATE","EVENT_CLICK","SELECTOR_TARGET_LINKS","SELECTOR_NAV_LINKS","SELECTOR_LINK_ITEMS","rootMargin","smoothScroll","threshold","ScrollSpy","_targetLinks","_observableSections","_rootElement","_activeTarget","_observer","_previousScrollData","visibleEntryTop","parentScrollTop","refresh","_initializeTargetsAndObservables","_maybeEnableSmoothScroll","disconnect","_getNewObserver","section","observe","observableSection","scrollTo","behavior","IntersectionObserver","_observerCallback","targetElement","_process","userScrollsDown","isIntersecting","_clearActiveClass","entryIsLowerThanPrevious","targetLinks","anchor","decodeURI","_activateParents","listGroup","activeNodes","spy","ARROW_LEFT_KEY","ARROW_RIGHT_KEY","HOME_KEY","END_KEY","NOT_SELECTOR_DROPDOWN_TOGGLE","SELECTOR_INNER_ELEM","SELECTOR_DATA_TOGGLE_ACTIVE","Tab","_setInitialAttributes","_getChildren","innerElem","_elemIsActive","active","_getActiveElem","hideEvent","_deactivate","_activate","relatedElem","_toggleDropDown","nextActiveElement","preventScroll","_setAttributeIfNotExists","_setInitialAttributesOnChild","_getInnerElement","isActive","outerElem","_getOuterElement","_setInitialAttributesOnTargetPanel","open","EVENT_MOUSEOVER","EVENT_MOUSEOUT","EVENT_FOCUSOUT","CLASS_NAME_HIDE","autohide","Toast","_hasMouseInteraction","_hasKeyboardInteraction","_clearTimeout","_maybeScheduleHide","isShown","_onInteraction","isInteracting"],"sources":["../../js/src/dom/data.js","../../js/src/util/index.js","../../js/src/dom/event-handler.js","../../js/src/dom/manipulator.js","../../js/src/util/config.js","../../js/src/base-component.js","../../js/src/dom/selector-engine.js","../../js/src/util/component-functions.js","../../js/src/alert.js","../../js/src/button.js","../../js/src/util/swipe.js","../../js/src/carousel.js","../../js/src/collapse.js","../../node_modules/@popperjs/core/lib/enums.js","../../node_modules/@popperjs/core/lib/dom-utils/getNodeName.js","../../node_modules/@popperjs/core/lib/dom-utils/getWindow.js","../../node_modules/@popperjs/core/lib/dom-utils/instanceOf.js","../../node_modules/@popperjs/core/lib/modifiers/applyStyles.js","../../node_modules/@popperjs/core/lib/utils/getBasePlacement.js","../../node_modules/@popperjs/core/lib/utils/math.js","../../node_modules/@popperjs/core/lib/utils/userAgent.js","../../node_modules/@popperjs/core/lib/dom-utils/isLayoutViewport.js","../../node_modules/@popperjs/core/lib/dom-utils/getBoundingClientRect.js","../../node_modules/@popperjs/core/lib/dom-utils/getLayoutRect.js","../../node_modules/@popperjs/core/lib/dom-utils/contains.js","../../node_modules/@popperjs/core/lib/dom-utils/getComputedStyle.js","../../node_modules/@popperjs/core/lib/dom-utils/isTableElement.js","../../node_modules/@popperjs/core/lib/dom-utils/getDocumentElement.js","../../node_modules/@popperjs/core/lib/dom-utils/getParentNode.js","../../node_modules/@popperjs/core/lib/dom-utils/getOffsetParent.js","../../node_modules/@popperjs/core/lib/utils/getMainAxisFromPlacement.js","../../node_modules/@popperjs/core/lib/utils/within.js","../../node_modules/@popperjs/core/lib/utils/mergePaddingObject.js","../../node_modules/@popperjs/core/lib/utils/getFreshSideObject.js","../../node_modules/@popperjs/core/lib/utils/expandToHashMap.js","../../node_modules/@popperjs/core/lib/modifiers/arrow.js","../../node_modules/@popperjs/core/lib/utils/getVariation.js","../../node_modules/@popperjs/core/lib/modifiers/computeStyles.js","../../node_modules/@popperjs/core/lib/modifiers/eventListeners.js","../../node_modules/@popperjs/core/lib/utils/getOppositePlacement.js","../../node_modules/@popperjs/core/lib/utils/getOppositeVariationPlacement.js","../../node_modules/@popperjs/core/lib/dom-utils/getWindowScroll.js","../../node_modules/@popperjs/core/lib/dom-utils/getWindowScrollBarX.js","../../node_modules/@popperjs/core/lib/dom-utils/isScrollParent.js","../../node_modules/@popperjs/core/lib/dom-utils/getScrollParent.js","../../node_modules/@popperjs/core/lib/dom-utils/listScrollParents.js","../../node_modules/@popperjs/core/lib/utils/rectToClientRect.js","../../node_modules/@popperjs/core/lib/dom-utils/getClippingRect.js","../../node_modules/@popperjs/core/lib/dom-utils/getViewportRect.js","../../node_modules/@popperjs/core/lib/dom-utils/getDocumentRect.js","../../node_modules/@popperjs/core/lib/utils/computeOffsets.js","../../node_modules/@popperjs/core/lib/utils/detectOverflow.js","../../node_modules/@popperjs/core/lib/utils/computeAutoPlacement.js","../../node_modules/@popperjs/core/lib/modifiers/flip.js","../../node_modules/@popperjs/core/lib/modifiers/hide.js","../../node_modules/@popperjs/core/lib/modifiers/offset.js","../../node_modules/@popperjs/core/lib/modifiers/popperOffsets.js","../../node_modules/@popperjs/core/lib/modifiers/preventOverflow.js","../../node_modules/@popperjs/core/lib/utils/getAltAxis.js","../../node_modules/@popperjs/core/lib/dom-utils/getCompositeRect.js","../../node_modules/@popperjs/core/lib/dom-utils/getNodeScroll.js","../../node_modules/@popperjs/core/lib/dom-utils/getHTMLElementScroll.js","../../node_modules/@popperjs/core/lib/utils/orderModifiers.js","../../node_modules/@popperjs/core/lib/createPopper.js","../../node_modules/@popperjs/core/lib/utils/debounce.js","../../node_modules/@popperjs/core/lib/utils/mergeByName.js","../../node_modules/@popperjs/core/lib/popper-lite.js","../../node_modules/@popperjs/core/lib/popper.js","../../js/src/dropdown.js","../../js/src/util/backdrop.js","../../js/src/util/focustrap.js","../../js/src/util/scrollbar.js","../../js/src/modal.js","../../js/src/offcanvas.js","../../js/src/util/sanitizer.js","../../js/src/util/template-factory.js","../../js/src/tooltip.js","../../js/src/popover.js","../../js/src/scrollspy.js","../../js/src/tab.js","../../js/src/toast.js","../../js/index.umd.js"],"sourcesContent":["/**\n * --------------------------------------------------------------------------\n * Bootstrap dom/data.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\n/**\n * Constants\n */\n\nconst elementMap = new Map()\n\nexport default {\n set(element, key, instance) {\n if (!elementMap.has(element)) {\n elementMap.set(element, new Map())\n }\n\n const instanceMap = elementMap.get(element)\n\n // make it clear we only want one instance per element\n // can be removed later when multiple key/instances are fine to be used\n if (!instanceMap.has(key) && instanceMap.size !== 0) {\n // eslint-disable-next-line no-console\n console.error(`Bootstrap doesn't allow more than one instance per element. Bound instance: ${Array.from(instanceMap.keys())[0]}.`)\n return\n }\n\n instanceMap.set(key, instance)\n },\n\n get(element, key) {\n if (elementMap.has(element)) {\n return elementMap.get(element).get(key) || null\n }\n\n return null\n },\n\n remove(element, key) {\n if (!elementMap.has(element)) {\n return\n }\n\n const instanceMap = elementMap.get(element)\n\n instanceMap.delete(key)\n\n // free up element references if there are no instances left for an element\n if (instanceMap.size === 0) {\n elementMap.delete(element)\n }\n }\n}\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/index.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nconst MAX_UID = 1_000_000\nconst MILLISECONDS_MULTIPLIER = 1000\nconst TRANSITION_END = 'transitionend'\n\n/**\n * Properly escape IDs selectors to handle weird IDs\n * @param {string} selector\n * @returns {string}\n */\nconst parseSelector = selector => {\n if (selector && window.CSS && window.CSS.escape) {\n // document.querySelector needs escaping to handle IDs (html5+) containing for instance /\n selector = selector.replace(/#([^\\s\"#']+)/g, (match, id) => `#${CSS.escape(id)}`)\n }\n\n return selector\n}\n\n// Shout-out Angus Croll (https://goo.gl/pxwQGp)\nconst toType = object => {\n if (object === null || object === undefined) {\n return `${object}`\n }\n\n return Object.prototype.toString.call(object).match(/\\s([a-z]+)/i)[1].toLowerCase()\n}\n\n/**\n * Public Util API\n */\n\nconst getUID = prefix => {\n do {\n prefix += Math.floor(Math.random() * MAX_UID)\n } while (document.getElementById(prefix))\n\n return prefix\n}\n\nconst getTransitionDurationFromElement = element => {\n if (!element) {\n return 0\n }\n\n // Get transition-duration of the element\n let { transitionDuration, transitionDelay } = window.getComputedStyle(element)\n\n const floatTransitionDuration = Number.parseFloat(transitionDuration)\n const floatTransitionDelay = Number.parseFloat(transitionDelay)\n\n // Return 0 if element or transition duration is not found\n if (!floatTransitionDuration && !floatTransitionDelay) {\n return 0\n }\n\n // If multiple durations are defined, take the first\n transitionDuration = transitionDuration.split(',')[0]\n transitionDelay = transitionDelay.split(',')[0]\n\n return (Number.parseFloat(transitionDuration) + Number.parseFloat(transitionDelay)) * MILLISECONDS_MULTIPLIER\n}\n\nconst triggerTransitionEnd = element => {\n element.dispatchEvent(new Event(TRANSITION_END))\n}\n\nconst isElement = object => {\n if (!object || typeof object !== 'object') {\n return false\n }\n\n if (typeof object.jquery !== 'undefined') {\n object = object[0]\n }\n\n return typeof object.nodeType !== 'undefined'\n}\n\nconst getElement = object => {\n // it's a jQuery object or a node element\n if (isElement(object)) {\n return object.jquery ? object[0] : object\n }\n\n if (typeof object === 'string' && object.length > 0) {\n return document.querySelector(parseSelector(object))\n }\n\n return null\n}\n\nconst isVisible = element => {\n if (!isElement(element) || element.getClientRects().length === 0) {\n return false\n }\n\n const elementIsVisible = getComputedStyle(element).getPropertyValue('visibility') === 'visible'\n // Handle `details` element as its content may falsie appear visible when it is closed\n const closedDetails = element.closest('details:not([open])')\n\n if (!closedDetails) {\n return elementIsVisible\n }\n\n if (closedDetails !== element) {\n const summary = element.closest('summary')\n if (summary && summary.parentNode !== closedDetails) {\n return false\n }\n\n if (summary === null) {\n return false\n }\n }\n\n return elementIsVisible\n}\n\nconst isDisabled = element => {\n if (!element || element.nodeType !== Node.ELEMENT_NODE) {\n return true\n }\n\n if (element.classList.contains('disabled')) {\n return true\n }\n\n if (typeof element.disabled !== 'undefined') {\n return element.disabled\n }\n\n return element.hasAttribute('disabled') && element.getAttribute('disabled') !== 'false'\n}\n\nconst findShadowRoot = element => {\n if (!document.documentElement.attachShadow) {\n return null\n }\n\n // Can find the shadow root otherwise it'll return the document\n if (typeof element.getRootNode === 'function') {\n const root = element.getRootNode()\n return root instanceof ShadowRoot ? root : null\n }\n\n if (element instanceof ShadowRoot) {\n return element\n }\n\n // when we don't find a shadow root\n if (!element.parentNode) {\n return null\n }\n\n return findShadowRoot(element.parentNode)\n}\n\nconst noop = () => {}\n\n/**\n * Trick to restart an element's animation\n *\n * @param {HTMLElement} element\n * @return void\n *\n * @see https://www.charistheo.io/blog/2021/02/restart-a-css-animation-with-javascript/#restarting-a-css-animation\n */\nconst reflow = element => {\n element.offsetHeight // eslint-disable-line no-unused-expressions\n}\n\nconst getjQuery = () => {\n if (window.jQuery && !document.body.hasAttribute('data-bs-no-jquery')) {\n return window.jQuery\n }\n\n return null\n}\n\nconst DOMContentLoadedCallbacks = []\n\nconst onDOMContentLoaded = callback => {\n if (document.readyState === 'loading') {\n // add listener on the first call when the document is in loading state\n if (!DOMContentLoadedCallbacks.length) {\n document.addEventListener('DOMContentLoaded', () => {\n for (const callback of DOMContentLoadedCallbacks) {\n callback()\n }\n })\n }\n\n DOMContentLoadedCallbacks.push(callback)\n } else {\n callback()\n }\n}\n\nconst isRTL = () => document.documentElement.dir === 'rtl'\n\nconst defineJQueryPlugin = plugin => {\n onDOMContentLoaded(() => {\n const $ = getjQuery()\n /* istanbul ignore if */\n if ($) {\n const name = plugin.NAME\n const JQUERY_NO_CONFLICT = $.fn[name]\n $.fn[name] = plugin.jQueryInterface\n $.fn[name].Constructor = plugin\n $.fn[name].noConflict = () => {\n $.fn[name] = JQUERY_NO_CONFLICT\n return plugin.jQueryInterface\n }\n }\n })\n}\n\nconst execute = (possibleCallback, args = [], defaultValue = possibleCallback) => {\n return typeof possibleCallback === 'function' ? possibleCallback(...args) : defaultValue\n}\n\nconst executeAfterTransition = (callback, transitionElement, waitForTransition = true) => {\n if (!waitForTransition) {\n execute(callback)\n return\n }\n\n const durationPadding = 5\n const emulatedDuration = getTransitionDurationFromElement(transitionElement) + durationPadding\n\n let called = false\n\n const handler = ({ target }) => {\n if (target !== transitionElement) {\n return\n }\n\n called = true\n transitionElement.removeEventListener(TRANSITION_END, handler)\n execute(callback)\n }\n\n transitionElement.addEventListener(TRANSITION_END, handler)\n setTimeout(() => {\n if (!called) {\n triggerTransitionEnd(transitionElement)\n }\n }, emulatedDuration)\n}\n\n/**\n * Return the previous/next element of a list.\n *\n * @param {array} list The list of elements\n * @param activeElement The active element\n * @param shouldGetNext Choose to get next or previous element\n * @param isCycleAllowed\n * @return {Element|elem} The proper element\n */\nconst getNextActiveElement = (list, activeElement, shouldGetNext, isCycleAllowed) => {\n const listLength = list.length\n let index = list.indexOf(activeElement)\n\n // if the element does not exist in the list return an element\n // depending on the direction and if cycle is allowed\n if (index === -1) {\n return !shouldGetNext && isCycleAllowed ? list[listLength - 1] : list[0]\n }\n\n index += shouldGetNext ? 1 : -1\n\n if (isCycleAllowed) {\n index = (index + listLength) % listLength\n }\n\n return list[Math.max(0, Math.min(index, listLength - 1))]\n}\n\nexport {\n defineJQueryPlugin,\n execute,\n executeAfterTransition,\n findShadowRoot,\n getElement,\n getjQuery,\n getNextActiveElement,\n getTransitionDurationFromElement,\n getUID,\n isDisabled,\n isElement,\n isRTL,\n isVisible,\n noop,\n onDOMContentLoaded,\n parseSelector,\n reflow,\n triggerTransitionEnd,\n toType\n}\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap dom/event-handler.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport { getjQuery } from '../util/index.js'\n\n/**\n * Constants\n */\n\nconst namespaceRegex = /[^.]*(?=\\..*)\\.|.*/\nconst stripNameRegex = /\\..*/\nconst stripUidRegex = /::\\d+$/\nconst eventRegistry = {} // Events storage\nlet uidEvent = 1\nconst customEvents = {\n mouseenter: 'mouseover',\n mouseleave: 'mouseout'\n}\n\nconst nativeEvents = new Set([\n 'click',\n 'dblclick',\n 'mouseup',\n 'mousedown',\n 'contextmenu',\n 'mousewheel',\n 'DOMMouseScroll',\n 'mouseover',\n 'mouseout',\n 'mousemove',\n 'selectstart',\n 'selectend',\n 'keydown',\n 'keypress',\n 'keyup',\n 'orientationchange',\n 'touchstart',\n 'touchmove',\n 'touchend',\n 'touchcancel',\n 'pointerdown',\n 'pointermove',\n 'pointerup',\n 'pointerleave',\n 'pointercancel',\n 'gesturestart',\n 'gesturechange',\n 'gestureend',\n 'focus',\n 'blur',\n 'change',\n 'reset',\n 'select',\n 'submit',\n 'focusin',\n 'focusout',\n 'load',\n 'unload',\n 'beforeunload',\n 'resize',\n 'move',\n 'DOMContentLoaded',\n 'readystatechange',\n 'error',\n 'abort',\n 'scroll'\n])\n\n/**\n * Private methods\n */\n\nfunction makeEventUid(element, uid) {\n return (uid && `${uid}::${uidEvent++}`) || element.uidEvent || uidEvent++\n}\n\nfunction getElementEvents(element) {\n const uid = makeEventUid(element)\n\n element.uidEvent = uid\n eventRegistry[uid] = eventRegistry[uid] || {}\n\n return eventRegistry[uid]\n}\n\nfunction bootstrapHandler(element, fn) {\n return function handler(event) {\n hydrateObj(event, { delegateTarget: element })\n\n if (handler.oneOff) {\n EventHandler.off(element, event.type, fn)\n }\n\n return fn.apply(element, [event])\n }\n}\n\nfunction bootstrapDelegationHandler(element, selector, fn) {\n return function handler(event) {\n const domElements = element.querySelectorAll(selector)\n\n for (let { target } = event; target && target !== this; target = target.parentNode) {\n for (const domElement of domElements) {\n if (domElement !== target) {\n continue\n }\n\n hydrateObj(event, { delegateTarget: target })\n\n if (handler.oneOff) {\n EventHandler.off(element, event.type, selector, fn)\n }\n\n return fn.apply(target, [event])\n }\n }\n }\n}\n\nfunction findHandler(events, callable, delegationSelector = null) {\n return Object.values(events)\n .find(event => event.callable === callable && event.delegationSelector === delegationSelector)\n}\n\nfunction normalizeParameters(originalTypeEvent, handler, delegationFunction) {\n const isDelegated = typeof handler === 'string'\n // TODO: tooltip passes `false` instead of selector, so we need to check\n const callable = isDelegated ? delegationFunction : (handler || delegationFunction)\n let typeEvent = getTypeEvent(originalTypeEvent)\n\n if (!nativeEvents.has(typeEvent)) {\n typeEvent = originalTypeEvent\n }\n\n return [isDelegated, callable, typeEvent]\n}\n\nfunction addHandler(element, originalTypeEvent, handler, delegationFunction, oneOff) {\n if (typeof originalTypeEvent !== 'string' || !element) {\n return\n }\n\n let [isDelegated, callable, typeEvent] = normalizeParameters(originalTypeEvent, handler, delegationFunction)\n\n // in case of mouseenter or mouseleave wrap the handler within a function that checks for its DOM position\n // this prevents the handler from being dispatched the same way as mouseover or mouseout does\n if (originalTypeEvent in customEvents) {\n const wrapFunction = fn => {\n return function (event) {\n if (!event.relatedTarget || (event.relatedTarget !== event.delegateTarget && !event.delegateTarget.contains(event.relatedTarget))) {\n return fn.call(this, event)\n }\n }\n }\n\n callable = wrapFunction(callable)\n }\n\n const events = getElementEvents(element)\n const handlers = events[typeEvent] || (events[typeEvent] = {})\n const previousFunction = findHandler(handlers, callable, isDelegated ? handler : null)\n\n if (previousFunction) {\n previousFunction.oneOff = previousFunction.oneOff && oneOff\n\n return\n }\n\n const uid = makeEventUid(callable, originalTypeEvent.replace(namespaceRegex, ''))\n const fn = isDelegated ?\n bootstrapDelegationHandler(element, handler, callable) :\n bootstrapHandler(element, callable)\n\n fn.delegationSelector = isDelegated ? handler : null\n fn.callable = callable\n fn.oneOff = oneOff\n fn.uidEvent = uid\n handlers[uid] = fn\n\n element.addEventListener(typeEvent, fn, isDelegated)\n}\n\nfunction removeHandler(element, events, typeEvent, handler, delegationSelector) {\n const fn = findHandler(events[typeEvent], handler, delegationSelector)\n\n if (!fn) {\n return\n }\n\n element.removeEventListener(typeEvent, fn, Boolean(delegationSelector))\n delete events[typeEvent][fn.uidEvent]\n}\n\nfunction removeNamespacedHandlers(element, events, typeEvent, namespace) {\n const storeElementEvent = events[typeEvent] || {}\n\n for (const [handlerKey, event] of Object.entries(storeElementEvent)) {\n if (handlerKey.includes(namespace)) {\n removeHandler(element, events, typeEvent, event.callable, event.delegationSelector)\n }\n }\n}\n\nfunction getTypeEvent(event) {\n // allow to get the native events from namespaced events ('click.bs.button' --> 'click')\n event = event.replace(stripNameRegex, '')\n return customEvents[event] || event\n}\n\nconst EventHandler = {\n on(element, event, handler, delegationFunction) {\n addHandler(element, event, handler, delegationFunction, false)\n },\n\n one(element, event, handler, delegationFunction) {\n addHandler(element, event, handler, delegationFunction, true)\n },\n\n off(element, originalTypeEvent, handler, delegationFunction) {\n if (typeof originalTypeEvent !== 'string' || !element) {\n return\n }\n\n const [isDelegated, callable, typeEvent] = normalizeParameters(originalTypeEvent, handler, delegationFunction)\n const inNamespace = typeEvent !== originalTypeEvent\n const events = getElementEvents(element)\n const storeElementEvent = events[typeEvent] || {}\n const isNamespace = originalTypeEvent.startsWith('.')\n\n if (typeof callable !== 'undefined') {\n // Simplest case: handler is passed, remove that listener ONLY.\n if (!Object.keys(storeElementEvent).length) {\n return\n }\n\n removeHandler(element, events, typeEvent, callable, isDelegated ? handler : null)\n return\n }\n\n if (isNamespace) {\n for (const elementEvent of Object.keys(events)) {\n removeNamespacedHandlers(element, events, elementEvent, originalTypeEvent.slice(1))\n }\n }\n\n for (const [keyHandlers, event] of Object.entries(storeElementEvent)) {\n const handlerKey = keyHandlers.replace(stripUidRegex, '')\n\n if (!inNamespace || originalTypeEvent.includes(handlerKey)) {\n removeHandler(element, events, typeEvent, event.callable, event.delegationSelector)\n }\n }\n },\n\n trigger(element, event, args) {\n if (typeof event !== 'string' || !element) {\n return null\n }\n\n const $ = getjQuery()\n const typeEvent = getTypeEvent(event)\n const inNamespace = event !== typeEvent\n\n let jQueryEvent = null\n let bubbles = true\n let nativeDispatch = true\n let defaultPrevented = false\n\n if (inNamespace && $) {\n jQueryEvent = $.Event(event, args)\n\n $(element).trigger(jQueryEvent)\n bubbles = !jQueryEvent.isPropagationStopped()\n nativeDispatch = !jQueryEvent.isImmediatePropagationStopped()\n defaultPrevented = jQueryEvent.isDefaultPrevented()\n }\n\n const evt = hydrateObj(new Event(event, { bubbles, cancelable: true }), args)\n\n if (defaultPrevented) {\n evt.preventDefault()\n }\n\n if (nativeDispatch) {\n element.dispatchEvent(evt)\n }\n\n if (evt.defaultPrevented && jQueryEvent) {\n jQueryEvent.preventDefault()\n }\n\n return evt\n }\n}\n\nfunction hydrateObj(obj, meta = {}) {\n for (const [key, value] of Object.entries(meta)) {\n try {\n obj[key] = value\n } catch {\n Object.defineProperty(obj, key, {\n configurable: true,\n get() {\n return value\n }\n })\n }\n }\n\n return obj\n}\n\nexport default EventHandler\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap dom/manipulator.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nfunction normalizeData(value) {\n if (value === 'true') {\n return true\n }\n\n if (value === 'false') {\n return false\n }\n\n if (value === Number(value).toString()) {\n return Number(value)\n }\n\n if (value === '' || value === 'null') {\n return null\n }\n\n if (typeof value !== 'string') {\n return value\n }\n\n try {\n return JSON.parse(decodeURIComponent(value))\n } catch {\n return value\n }\n}\n\nfunction normalizeDataKey(key) {\n return key.replace(/[A-Z]/g, chr => `-${chr.toLowerCase()}`)\n}\n\nconst Manipulator = {\n setDataAttribute(element, key, value) {\n element.setAttribute(`data-bs-${normalizeDataKey(key)}`, value)\n },\n\n removeDataAttribute(element, key) {\n element.removeAttribute(`data-bs-${normalizeDataKey(key)}`)\n },\n\n getDataAttributes(element) {\n if (!element) {\n return {}\n }\n\n const attributes = {}\n const bsKeys = Object.keys(element.dataset).filter(key => key.startsWith('bs') && !key.startsWith('bsConfig'))\n\n for (const key of bsKeys) {\n let pureKey = key.replace(/^bs/, '')\n pureKey = pureKey.charAt(0).toLowerCase() + pureKey.slice(1, pureKey.length)\n attributes[pureKey] = normalizeData(element.dataset[key])\n }\n\n return attributes\n },\n\n getDataAttribute(element, key) {\n return normalizeData(element.getAttribute(`data-bs-${normalizeDataKey(key)}`))\n }\n}\n\nexport default Manipulator\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/config.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport Manipulator from '../dom/manipulator.js'\nimport { isElement, toType } from './index.js'\n\n/**\n * Class definition\n */\n\nclass Config {\n // Getters\n static get Default() {\n return {}\n }\n\n static get DefaultType() {\n return {}\n }\n\n static get NAME() {\n throw new Error('You have to implement the static method \"NAME\", for each component!')\n }\n\n _getConfig(config) {\n config = this._mergeConfigObj(config)\n config = this._configAfterMerge(config)\n this._typeCheckConfig(config)\n return config\n }\n\n _configAfterMerge(config) {\n return config\n }\n\n _mergeConfigObj(config, element) {\n const jsonConfig = isElement(element) ? Manipulator.getDataAttribute(element, 'config') : {} // try to parse\n\n return {\n ...this.constructor.Default,\n ...(typeof jsonConfig === 'object' ? jsonConfig : {}),\n ...(isElement(element) ? Manipulator.getDataAttributes(element) : {}),\n ...(typeof config === 'object' ? config : {})\n }\n }\n\n _typeCheckConfig(config, configTypes = this.constructor.DefaultType) {\n for (const [property, expectedTypes] of Object.entries(configTypes)) {\n const value = config[property]\n const valueType = isElement(value) ? 'element' : toType(value)\n\n if (!new RegExp(expectedTypes).test(valueType)) {\n throw new TypeError(\n `${this.constructor.NAME.toUpperCase()}: Option \"${property}\" provided type \"${valueType}\" but expected type \"${expectedTypes}\".`\n )\n }\n }\n }\n}\n\nexport default Config\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap base-component.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport Data from './dom/data.js'\nimport EventHandler from './dom/event-handler.js'\nimport Config from './util/config.js'\nimport { executeAfterTransition, getElement } from './util/index.js'\n\n/**\n * Constants\n */\n\nconst VERSION = '5.3.1'\n\n/**\n * Class definition\n */\n\nclass BaseComponent extends Config {\n constructor(element, config) {\n super()\n\n element = getElement(element)\n if (!element) {\n return\n }\n\n this._element = element\n this._config = this._getConfig(config)\n\n Data.set(this._element, this.constructor.DATA_KEY, this)\n }\n\n // Public\n dispose() {\n Data.remove(this._element, this.constructor.DATA_KEY)\n EventHandler.off(this._element, this.constructor.EVENT_KEY)\n\n for (const propertyName of Object.getOwnPropertyNames(this)) {\n this[propertyName] = null\n }\n }\n\n _queueCallback(callback, element, isAnimated = true) {\n executeAfterTransition(callback, element, isAnimated)\n }\n\n _getConfig(config) {\n config = this._mergeConfigObj(config, this._element)\n config = this._configAfterMerge(config)\n this._typeCheckConfig(config)\n return config\n }\n\n // Static\n static getInstance(element) {\n return Data.get(getElement(element), this.DATA_KEY)\n }\n\n static getOrCreateInstance(element, config = {}) {\n return this.getInstance(element) || new this(element, typeof config === 'object' ? config : null)\n }\n\n static get VERSION() {\n return VERSION\n }\n\n static get DATA_KEY() {\n return `bs.${this.NAME}`\n }\n\n static get EVENT_KEY() {\n return `.${this.DATA_KEY}`\n }\n\n static eventName(name) {\n return `${name}${this.EVENT_KEY}`\n }\n}\n\nexport default BaseComponent\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap dom/selector-engine.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport { isDisabled, isVisible, parseSelector } from '../util/index.js'\n\nconst getSelector = element => {\n let selector = element.getAttribute('data-bs-target')\n\n if (!selector || selector === '#') {\n let hrefAttribute = element.getAttribute('href')\n\n // The only valid content that could double as a selector are IDs or classes,\n // so everything starting with `#` or `.`. If a \"real\" URL is used as the selector,\n // `document.querySelector` will rightfully complain it is invalid.\n // See https://github.com/twbs/bootstrap/issues/32273\n if (!hrefAttribute || (!hrefAttribute.includes('#') && !hrefAttribute.startsWith('.'))) {\n return null\n }\n\n // Just in case some CMS puts out a full URL with the anchor appended\n if (hrefAttribute.includes('#') && !hrefAttribute.startsWith('#')) {\n hrefAttribute = `#${hrefAttribute.split('#')[1]}`\n }\n\n selector = hrefAttribute && hrefAttribute !== '#' ? hrefAttribute.trim() : null\n }\n\n return parseSelector(selector)\n}\n\nconst SelectorEngine = {\n find(selector, element = document.documentElement) {\n return [].concat(...Element.prototype.querySelectorAll.call(element, selector))\n },\n\n findOne(selector, element = document.documentElement) {\n return Element.prototype.querySelector.call(element, selector)\n },\n\n children(element, selector) {\n return [].concat(...element.children).filter(child => child.matches(selector))\n },\n\n parents(element, selector) {\n const parents = []\n let ancestor = element.parentNode.closest(selector)\n\n while (ancestor) {\n parents.push(ancestor)\n ancestor = ancestor.parentNode.closest(selector)\n }\n\n return parents\n },\n\n prev(element, selector) {\n let previous = element.previousElementSibling\n\n while (previous) {\n if (previous.matches(selector)) {\n return [previous]\n }\n\n previous = previous.previousElementSibling\n }\n\n return []\n },\n // TODO: this is now unused; remove later along with prev()\n next(element, selector) {\n let next = element.nextElementSibling\n\n while (next) {\n if (next.matches(selector)) {\n return [next]\n }\n\n next = next.nextElementSibling\n }\n\n return []\n },\n\n focusableChildren(element) {\n const focusables = [\n 'a',\n 'button',\n 'input',\n 'textarea',\n 'select',\n 'details',\n '[tabindex]',\n '[contenteditable=\"true\"]'\n ].map(selector => `${selector}:not([tabindex^=\"-\"])`).join(',')\n\n return this.find(focusables, element).filter(el => !isDisabled(el) && isVisible(el))\n },\n\n getSelectorFromElement(element) {\n const selector = getSelector(element)\n\n if (selector) {\n return SelectorEngine.findOne(selector) ? selector : null\n }\n\n return null\n },\n\n getElementFromSelector(element) {\n const selector = getSelector(element)\n\n return selector ? SelectorEngine.findOne(selector) : null\n },\n\n getMultipleElementsFromSelector(element) {\n const selector = getSelector(element)\n\n return selector ? SelectorEngine.find(selector) : []\n }\n}\n\nexport default SelectorEngine\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/component-functions.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport EventHandler from '../dom/event-handler.js'\nimport SelectorEngine from '../dom/selector-engine.js'\nimport { isDisabled } from './index.js'\n\nconst enableDismissTrigger = (component, method = 'hide') => {\n const clickEvent = `click.dismiss${component.EVENT_KEY}`\n const name = component.NAME\n\n EventHandler.on(document, clickEvent, `[data-bs-dismiss=\"${name}\"]`, function (event) {\n if (['A', 'AREA'].includes(this.tagName)) {\n event.preventDefault()\n }\n\n if (isDisabled(this)) {\n return\n }\n\n const target = SelectorEngine.getElementFromSelector(this) || this.closest(`.${name}`)\n const instance = component.getOrCreateInstance(target)\n\n // Method argument is left, for Alert and only, as it doesn't implement the 'hide' method\n instance[method]()\n })\n}\n\nexport {\n enableDismissTrigger\n}\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap alert.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport { enableDismissTrigger } from './util/component-functions.js'\nimport { defineJQueryPlugin } from './util/index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'alert'\nconst DATA_KEY = 'bs.alert'\nconst EVENT_KEY = `.${DATA_KEY}`\n\nconst EVENT_CLOSE = `close${EVENT_KEY}`\nconst EVENT_CLOSED = `closed${EVENT_KEY}`\nconst CLASS_NAME_FADE = 'fade'\nconst CLASS_NAME_SHOW = 'show'\n\n/**\n * Class definition\n */\n\nclass Alert extends BaseComponent {\n // Getters\n static get NAME() {\n return NAME\n }\n\n // Public\n close() {\n const closeEvent = EventHandler.trigger(this._element, EVENT_CLOSE)\n\n if (closeEvent.defaultPrevented) {\n return\n }\n\n this._element.classList.remove(CLASS_NAME_SHOW)\n\n const isAnimated = this._element.classList.contains(CLASS_NAME_FADE)\n this._queueCallback(() => this._destroyElement(), this._element, isAnimated)\n }\n\n // Private\n _destroyElement() {\n this._element.remove()\n EventHandler.trigger(this._element, EVENT_CLOSED)\n this.dispose()\n }\n\n // Static\n static jQueryInterface(config) {\n return this.each(function () {\n const data = Alert.getOrCreateInstance(this)\n\n if (typeof config !== 'string') {\n return\n }\n\n if (data[config] === undefined || config.startsWith('_') || config === 'constructor') {\n throw new TypeError(`No method named \"${config}\"`)\n }\n\n data[config](this)\n })\n }\n}\n\n/**\n * Data API implementation\n */\n\nenableDismissTrigger(Alert, 'close')\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Alert)\n\nexport default Alert\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap button.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport { defineJQueryPlugin } from './util/index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'button'\nconst DATA_KEY = 'bs.button'\nconst EVENT_KEY = `.${DATA_KEY}`\nconst DATA_API_KEY = '.data-api'\n\nconst CLASS_NAME_ACTIVE = 'active'\nconst SELECTOR_DATA_TOGGLE = '[data-bs-toggle=\"button\"]'\nconst EVENT_CLICK_DATA_API = `click${EVENT_KEY}${DATA_API_KEY}`\n\n/**\n * Class definition\n */\n\nclass Button extends BaseComponent {\n // Getters\n static get NAME() {\n return NAME\n }\n\n // Public\n toggle() {\n // Toggle class and sync the `aria-pressed` attribute with the return value of the `.toggle()` method\n this._element.setAttribute('aria-pressed', this._element.classList.toggle(CLASS_NAME_ACTIVE))\n }\n\n // Static\n static jQueryInterface(config) {\n return this.each(function () {\n const data = Button.getOrCreateInstance(this)\n\n if (config === 'toggle') {\n data[config]()\n }\n })\n }\n}\n\n/**\n * Data API implementation\n */\n\nEventHandler.on(document, EVENT_CLICK_DATA_API, SELECTOR_DATA_TOGGLE, event => {\n event.preventDefault()\n\n const button = event.target.closest(SELECTOR_DATA_TOGGLE)\n const data = Button.getOrCreateInstance(button)\n\n data.toggle()\n})\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Button)\n\nexport default Button\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/swipe.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport EventHandler from '../dom/event-handler.js'\nimport Config from './config.js'\nimport { execute } from './index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'swipe'\nconst EVENT_KEY = '.bs.swipe'\nconst EVENT_TOUCHSTART = `touchstart${EVENT_KEY}`\nconst EVENT_TOUCHMOVE = `touchmove${EVENT_KEY}`\nconst EVENT_TOUCHEND = `touchend${EVENT_KEY}`\nconst EVENT_POINTERDOWN = `pointerdown${EVENT_KEY}`\nconst EVENT_POINTERUP = `pointerup${EVENT_KEY}`\nconst POINTER_TYPE_TOUCH = 'touch'\nconst POINTER_TYPE_PEN = 'pen'\nconst CLASS_NAME_POINTER_EVENT = 'pointer-event'\nconst SWIPE_THRESHOLD = 40\n\nconst Default = {\n endCallback: null,\n leftCallback: null,\n rightCallback: null\n}\n\nconst DefaultType = {\n endCallback: '(function|null)',\n leftCallback: '(function|null)',\n rightCallback: '(function|null)'\n}\n\n/**\n * Class definition\n */\n\nclass Swipe extends Config {\n constructor(element, config) {\n super()\n this._element = element\n\n if (!element || !Swipe.isSupported()) {\n return\n }\n\n this._config = this._getConfig(config)\n this._deltaX = 0\n this._supportPointerEvents = Boolean(window.PointerEvent)\n this._initEvents()\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n dispose() {\n EventHandler.off(this._element, EVENT_KEY)\n }\n\n // Private\n _start(event) {\n if (!this._supportPointerEvents) {\n this._deltaX = event.touches[0].clientX\n\n return\n }\n\n if (this._eventIsPointerPenTouch(event)) {\n this._deltaX = event.clientX\n }\n }\n\n _end(event) {\n if (this._eventIsPointerPenTouch(event)) {\n this._deltaX = event.clientX - this._deltaX\n }\n\n this._handleSwipe()\n execute(this._config.endCallback)\n }\n\n _move(event) {\n this._deltaX = event.touches && event.touches.length > 1 ?\n 0 :\n event.touches[0].clientX - this._deltaX\n }\n\n _handleSwipe() {\n const absDeltaX = Math.abs(this._deltaX)\n\n if (absDeltaX <= SWIPE_THRESHOLD) {\n return\n }\n\n const direction = absDeltaX / this._deltaX\n\n this._deltaX = 0\n\n if (!direction) {\n return\n }\n\n execute(direction > 0 ? this._config.rightCallback : this._config.leftCallback)\n }\n\n _initEvents() {\n if (this._supportPointerEvents) {\n EventHandler.on(this._element, EVENT_POINTERDOWN, event => this._start(event))\n EventHandler.on(this._element, EVENT_POINTERUP, event => this._end(event))\n\n this._element.classList.add(CLASS_NAME_POINTER_EVENT)\n } else {\n EventHandler.on(this._element, EVENT_TOUCHSTART, event => this._start(event))\n EventHandler.on(this._element, EVENT_TOUCHMOVE, event => this._move(event))\n EventHandler.on(this._element, EVENT_TOUCHEND, event => this._end(event))\n }\n }\n\n _eventIsPointerPenTouch(event) {\n return this._supportPointerEvents && (event.pointerType === POINTER_TYPE_PEN || event.pointerType === POINTER_TYPE_TOUCH)\n }\n\n // Static\n static isSupported() {\n return 'ontouchstart' in document.documentElement || navigator.maxTouchPoints > 0\n }\n}\n\nexport default Swipe\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap carousel.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport Manipulator from './dom/manipulator.js'\nimport SelectorEngine from './dom/selector-engine.js'\nimport {\n defineJQueryPlugin,\n getNextActiveElement,\n isRTL,\n isVisible,\n reflow,\n triggerTransitionEnd\n} from './util/index.js'\nimport Swipe from './util/swipe.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'carousel'\nconst DATA_KEY = 'bs.carousel'\nconst EVENT_KEY = `.${DATA_KEY}`\nconst DATA_API_KEY = '.data-api'\n\nconst ARROW_LEFT_KEY = 'ArrowLeft'\nconst ARROW_RIGHT_KEY = 'ArrowRight'\nconst TOUCHEVENT_COMPAT_WAIT = 500 // Time for mouse compat events to fire after touch\n\nconst ORDER_NEXT = 'next'\nconst ORDER_PREV = 'prev'\nconst DIRECTION_LEFT = 'left'\nconst DIRECTION_RIGHT = 'right'\n\nconst EVENT_SLIDE = `slide${EVENT_KEY}`\nconst EVENT_SLID = `slid${EVENT_KEY}`\nconst EVENT_KEYDOWN = `keydown${EVENT_KEY}`\nconst EVENT_MOUSEENTER = `mouseenter${EVENT_KEY}`\nconst EVENT_MOUSELEAVE = `mouseleave${EVENT_KEY}`\nconst EVENT_DRAG_START = `dragstart${EVENT_KEY}`\nconst EVENT_LOAD_DATA_API = `load${EVENT_KEY}${DATA_API_KEY}`\nconst EVENT_CLICK_DATA_API = `click${EVENT_KEY}${DATA_API_KEY}`\n\nconst CLASS_NAME_CAROUSEL = 'carousel'\nconst CLASS_NAME_ACTIVE = 'active'\nconst CLASS_NAME_SLIDE = 'slide'\nconst CLASS_NAME_END = 'carousel-item-end'\nconst CLASS_NAME_START = 'carousel-item-start'\nconst CLASS_NAME_NEXT = 'carousel-item-next'\nconst CLASS_NAME_PREV = 'carousel-item-prev'\n\nconst SELECTOR_ACTIVE = '.active'\nconst SELECTOR_ITEM = '.carousel-item'\nconst SELECTOR_ACTIVE_ITEM = SELECTOR_ACTIVE + SELECTOR_ITEM\nconst SELECTOR_ITEM_IMG = '.carousel-item img'\nconst SELECTOR_INDICATORS = '.carousel-indicators'\nconst SELECTOR_DATA_SLIDE = '[data-bs-slide], [data-bs-slide-to]'\nconst SELECTOR_DATA_RIDE = '[data-bs-ride=\"carousel\"]'\n\nconst KEY_TO_DIRECTION = {\n [ARROW_LEFT_KEY]: DIRECTION_RIGHT,\n [ARROW_RIGHT_KEY]: DIRECTION_LEFT\n}\n\nconst Default = {\n interval: 5000,\n keyboard: true,\n pause: 'hover',\n ride: false,\n touch: true,\n wrap: true\n}\n\nconst DefaultType = {\n interval: '(number|boolean)', // TODO:v6 remove boolean support\n keyboard: 'boolean',\n pause: '(string|boolean)',\n ride: '(boolean|string)',\n touch: 'boolean',\n wrap: 'boolean'\n}\n\n/**\n * Class definition\n */\n\nclass Carousel extends BaseComponent {\n constructor(element, config) {\n super(element, config)\n\n this._interval = null\n this._activeElement = null\n this._isSliding = false\n this.touchTimeout = null\n this._swipeHelper = null\n\n this._indicatorsElement = SelectorEngine.findOne(SELECTOR_INDICATORS, this._element)\n this._addEventListeners()\n\n if (this._config.ride === CLASS_NAME_CAROUSEL) {\n this.cycle()\n }\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n next() {\n this._slide(ORDER_NEXT)\n }\n\n nextWhenVisible() {\n // FIXME TODO use `document.visibilityState`\n // Don't call next when the page isn't visible\n // or the carousel or its parent isn't visible\n if (!document.hidden && isVisible(this._element)) {\n this.next()\n }\n }\n\n prev() {\n this._slide(ORDER_PREV)\n }\n\n pause() {\n if (this._isSliding) {\n triggerTransitionEnd(this._element)\n }\n\n this._clearInterval()\n }\n\n cycle() {\n this._clearInterval()\n this._updateInterval()\n\n this._interval = setInterval(() => this.nextWhenVisible(), this._config.interval)\n }\n\n _maybeEnableCycle() {\n if (!this._config.ride) {\n return\n }\n\n if (this._isSliding) {\n EventHandler.one(this._element, EVENT_SLID, () => this.cycle())\n return\n }\n\n this.cycle()\n }\n\n to(index) {\n const items = this._getItems()\n if (index > items.length - 1 || index < 0) {\n return\n }\n\n if (this._isSliding) {\n EventHandler.one(this._element, EVENT_SLID, () => this.to(index))\n return\n }\n\n const activeIndex = this._getItemIndex(this._getActive())\n if (activeIndex === index) {\n return\n }\n\n const order = index > activeIndex ? ORDER_NEXT : ORDER_PREV\n\n this._slide(order, items[index])\n }\n\n dispose() {\n if (this._swipeHelper) {\n this._swipeHelper.dispose()\n }\n\n super.dispose()\n }\n\n // Private\n _configAfterMerge(config) {\n config.defaultInterval = config.interval\n return config\n }\n\n _addEventListeners() {\n if (this._config.keyboard) {\n EventHandler.on(this._element, EVENT_KEYDOWN, event => this._keydown(event))\n }\n\n if (this._config.pause === 'hover') {\n EventHandler.on(this._element, EVENT_MOUSEENTER, () => this.pause())\n EventHandler.on(this._element, EVENT_MOUSELEAVE, () => this._maybeEnableCycle())\n }\n\n if (this._config.touch && Swipe.isSupported()) {\n this._addTouchEventListeners()\n }\n }\n\n _addTouchEventListeners() {\n for (const img of SelectorEngine.find(SELECTOR_ITEM_IMG, this._element)) {\n EventHandler.on(img, EVENT_DRAG_START, event => event.preventDefault())\n }\n\n const endCallBack = () => {\n if (this._config.pause !== 'hover') {\n return\n }\n\n // If it's a touch-enabled device, mouseenter/leave are fired as\n // part of the mouse compatibility events on first tap - the carousel\n // would stop cycling until user tapped out of it;\n // here, we listen for touchend, explicitly pause the carousel\n // (as if it's the second time we tap on it, mouseenter compat event\n // is NOT fired) and after a timeout (to allow for mouse compatibility\n // events to fire) we explicitly restart cycling\n\n this.pause()\n if (this.touchTimeout) {\n clearTimeout(this.touchTimeout)\n }\n\n this.touchTimeout = setTimeout(() => this._maybeEnableCycle(), TOUCHEVENT_COMPAT_WAIT + this._config.interval)\n }\n\n const swipeConfig = {\n leftCallback: () => this._slide(this._directionToOrder(DIRECTION_LEFT)),\n rightCallback: () => this._slide(this._directionToOrder(DIRECTION_RIGHT)),\n endCallback: endCallBack\n }\n\n this._swipeHelper = new Swipe(this._element, swipeConfig)\n }\n\n _keydown(event) {\n if (/input|textarea/i.test(event.target.tagName)) {\n return\n }\n\n const direction = KEY_TO_DIRECTION[event.key]\n if (direction) {\n event.preventDefault()\n this._slide(this._directionToOrder(direction))\n }\n }\n\n _getItemIndex(element) {\n return this._getItems().indexOf(element)\n }\n\n _setActiveIndicatorElement(index) {\n if (!this._indicatorsElement) {\n return\n }\n\n const activeIndicator = SelectorEngine.findOne(SELECTOR_ACTIVE, this._indicatorsElement)\n\n activeIndicator.classList.remove(CLASS_NAME_ACTIVE)\n activeIndicator.removeAttribute('aria-current')\n\n const newActiveIndicator = SelectorEngine.findOne(`[data-bs-slide-to=\"${index}\"]`, this._indicatorsElement)\n\n if (newActiveIndicator) {\n newActiveIndicator.classList.add(CLASS_NAME_ACTIVE)\n newActiveIndicator.setAttribute('aria-current', 'true')\n }\n }\n\n _updateInterval() {\n const element = this._activeElement || this._getActive()\n\n if (!element) {\n return\n }\n\n const elementInterval = Number.parseInt(element.getAttribute('data-bs-interval'), 10)\n\n this._config.interval = elementInterval || this._config.defaultInterval\n }\n\n _slide(order, element = null) {\n if (this._isSliding) {\n return\n }\n\n const activeElement = this._getActive()\n const isNext = order === ORDER_NEXT\n const nextElement = element || getNextActiveElement(this._getItems(), activeElement, isNext, this._config.wrap)\n\n if (nextElement === activeElement) {\n return\n }\n\n const nextElementIndex = this._getItemIndex(nextElement)\n\n const triggerEvent = eventName => {\n return EventHandler.trigger(this._element, eventName, {\n relatedTarget: nextElement,\n direction: this._orderToDirection(order),\n from: this._getItemIndex(activeElement),\n to: nextElementIndex\n })\n }\n\n const slideEvent = triggerEvent(EVENT_SLIDE)\n\n if (slideEvent.defaultPrevented) {\n return\n }\n\n if (!activeElement || !nextElement) {\n // Some weirdness is happening, so we bail\n // TODO: change tests that use empty divs to avoid this check\n return\n }\n\n const isCycling = Boolean(this._interval)\n this.pause()\n\n this._isSliding = true\n\n this._setActiveIndicatorElement(nextElementIndex)\n this._activeElement = nextElement\n\n const directionalClassName = isNext ? CLASS_NAME_START : CLASS_NAME_END\n const orderClassName = isNext ? CLASS_NAME_NEXT : CLASS_NAME_PREV\n\n nextElement.classList.add(orderClassName)\n\n reflow(nextElement)\n\n activeElement.classList.add(directionalClassName)\n nextElement.classList.add(directionalClassName)\n\n const completeCallBack = () => {\n nextElement.classList.remove(directionalClassName, orderClassName)\n nextElement.classList.add(CLASS_NAME_ACTIVE)\n\n activeElement.classList.remove(CLASS_NAME_ACTIVE, orderClassName, directionalClassName)\n\n this._isSliding = false\n\n triggerEvent(EVENT_SLID)\n }\n\n this._queueCallback(completeCallBack, activeElement, this._isAnimated())\n\n if (isCycling) {\n this.cycle()\n }\n }\n\n _isAnimated() {\n return this._element.classList.contains(CLASS_NAME_SLIDE)\n }\n\n _getActive() {\n return SelectorEngine.findOne(SELECTOR_ACTIVE_ITEM, this._element)\n }\n\n _getItems() {\n return SelectorEngine.find(SELECTOR_ITEM, this._element)\n }\n\n _clearInterval() {\n if (this._interval) {\n clearInterval(this._interval)\n this._interval = null\n }\n }\n\n _directionToOrder(direction) {\n if (isRTL()) {\n return direction === DIRECTION_LEFT ? ORDER_PREV : ORDER_NEXT\n }\n\n return direction === DIRECTION_LEFT ? ORDER_NEXT : ORDER_PREV\n }\n\n _orderToDirection(order) {\n if (isRTL()) {\n return order === ORDER_PREV ? DIRECTION_LEFT : DIRECTION_RIGHT\n }\n\n return order === ORDER_PREV ? DIRECTION_RIGHT : DIRECTION_LEFT\n }\n\n // Static\n static jQueryInterface(config) {\n return this.each(function () {\n const data = Carousel.getOrCreateInstance(this, config)\n\n if (typeof config === 'number') {\n data.to(config)\n return\n }\n\n if (typeof config === 'string') {\n if (data[config] === undefined || config.startsWith('_') || config === 'constructor') {\n throw new TypeError(`No method named \"${config}\"`)\n }\n\n data[config]()\n }\n })\n }\n}\n\n/**\n * Data API implementation\n */\n\nEventHandler.on(document, EVENT_CLICK_DATA_API, SELECTOR_DATA_SLIDE, function (event) {\n const target = SelectorEngine.getElementFromSelector(this)\n\n if (!target || !target.classList.contains(CLASS_NAME_CAROUSEL)) {\n return\n }\n\n event.preventDefault()\n\n const carousel = Carousel.getOrCreateInstance(target)\n const slideIndex = this.getAttribute('data-bs-slide-to')\n\n if (slideIndex) {\n carousel.to(slideIndex)\n carousel._maybeEnableCycle()\n return\n }\n\n if (Manipulator.getDataAttribute(this, 'slide') === 'next') {\n carousel.next()\n carousel._maybeEnableCycle()\n return\n }\n\n carousel.prev()\n carousel._maybeEnableCycle()\n})\n\nEventHandler.on(window, EVENT_LOAD_DATA_API, () => {\n const carousels = SelectorEngine.find(SELECTOR_DATA_RIDE)\n\n for (const carousel of carousels) {\n Carousel.getOrCreateInstance(carousel)\n }\n})\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Carousel)\n\nexport default Carousel\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap collapse.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport SelectorEngine from './dom/selector-engine.js'\nimport {\n defineJQueryPlugin,\n getElement,\n reflow\n} from './util/index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'collapse'\nconst DATA_KEY = 'bs.collapse'\nconst EVENT_KEY = `.${DATA_KEY}`\nconst DATA_API_KEY = '.data-api'\n\nconst EVENT_SHOW = `show${EVENT_KEY}`\nconst EVENT_SHOWN = `shown${EVENT_KEY}`\nconst EVENT_HIDE = `hide${EVENT_KEY}`\nconst EVENT_HIDDEN = `hidden${EVENT_KEY}`\nconst EVENT_CLICK_DATA_API = `click${EVENT_KEY}${DATA_API_KEY}`\n\nconst CLASS_NAME_SHOW = 'show'\nconst CLASS_NAME_COLLAPSE = 'collapse'\nconst CLASS_NAME_COLLAPSING = 'collapsing'\nconst CLASS_NAME_COLLAPSED = 'collapsed'\nconst CLASS_NAME_DEEPER_CHILDREN = `:scope .${CLASS_NAME_COLLAPSE} .${CLASS_NAME_COLLAPSE}`\nconst CLASS_NAME_HORIZONTAL = 'collapse-horizontal'\n\nconst WIDTH = 'width'\nconst HEIGHT = 'height'\n\nconst SELECTOR_ACTIVES = '.collapse.show, .collapse.collapsing'\nconst SELECTOR_DATA_TOGGLE = '[data-bs-toggle=\"collapse\"]'\n\nconst Default = {\n parent: null,\n toggle: true\n}\n\nconst DefaultType = {\n parent: '(null|element)',\n toggle: 'boolean'\n}\n\n/**\n * Class definition\n */\n\nclass Collapse extends BaseComponent {\n constructor(element, config) {\n super(element, config)\n\n this._isTransitioning = false\n this._triggerArray = []\n\n const toggleList = SelectorEngine.find(SELECTOR_DATA_TOGGLE)\n\n for (const elem of toggleList) {\n const selector = SelectorEngine.getSelectorFromElement(elem)\n const filterElement = SelectorEngine.find(selector)\n .filter(foundElement => foundElement === this._element)\n\n if (selector !== null && filterElement.length) {\n this._triggerArray.push(elem)\n }\n }\n\n this._initializeChildren()\n\n if (!this._config.parent) {\n this._addAriaAndCollapsedClass(this._triggerArray, this._isShown())\n }\n\n if (this._config.toggle) {\n this.toggle()\n }\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n toggle() {\n if (this._isShown()) {\n this.hide()\n } else {\n this.show()\n }\n }\n\n show() {\n if (this._isTransitioning || this._isShown()) {\n return\n }\n\n let activeChildren = []\n\n // find active children\n if (this._config.parent) {\n activeChildren = this._getFirstLevelChildren(SELECTOR_ACTIVES)\n .filter(element => element !== this._element)\n .map(element => Collapse.getOrCreateInstance(element, { toggle: false }))\n }\n\n if (activeChildren.length && activeChildren[0]._isTransitioning) {\n return\n }\n\n const startEvent = EventHandler.trigger(this._element, EVENT_SHOW)\n if (startEvent.defaultPrevented) {\n return\n }\n\n for (const activeInstance of activeChildren) {\n activeInstance.hide()\n }\n\n const dimension = this._getDimension()\n\n this._element.classList.remove(CLASS_NAME_COLLAPSE)\n this._element.classList.add(CLASS_NAME_COLLAPSING)\n\n this._element.style[dimension] = 0\n\n this._addAriaAndCollapsedClass(this._triggerArray, true)\n this._isTransitioning = true\n\n const complete = () => {\n this._isTransitioning = false\n\n this._element.classList.remove(CLASS_NAME_COLLAPSING)\n this._element.classList.add(CLASS_NAME_COLLAPSE, CLASS_NAME_SHOW)\n\n this._element.style[dimension] = ''\n\n EventHandler.trigger(this._element, EVENT_SHOWN)\n }\n\n const capitalizedDimension = dimension[0].toUpperCase() + dimension.slice(1)\n const scrollSize = `scroll${capitalizedDimension}`\n\n this._queueCallback(complete, this._element, true)\n this._element.style[dimension] = `${this._element[scrollSize]}px`\n }\n\n hide() {\n if (this._isTransitioning || !this._isShown()) {\n return\n }\n\n const startEvent = EventHandler.trigger(this._element, EVENT_HIDE)\n if (startEvent.defaultPrevented) {\n return\n }\n\n const dimension = this._getDimension()\n\n this._element.style[dimension] = `${this._element.getBoundingClientRect()[dimension]}px`\n\n reflow(this._element)\n\n this._element.classList.add(CLASS_NAME_COLLAPSING)\n this._element.classList.remove(CLASS_NAME_COLLAPSE, CLASS_NAME_SHOW)\n\n for (const trigger of this._triggerArray) {\n const element = SelectorEngine.getElementFromSelector(trigger)\n\n if (element && !this._isShown(element)) {\n this._addAriaAndCollapsedClass([trigger], false)\n }\n }\n\n this._isTransitioning = true\n\n const complete = () => {\n this._isTransitioning = false\n this._element.classList.remove(CLASS_NAME_COLLAPSING)\n this._element.classList.add(CLASS_NAME_COLLAPSE)\n EventHandler.trigger(this._element, EVENT_HIDDEN)\n }\n\n this._element.style[dimension] = ''\n\n this._queueCallback(complete, this._element, true)\n }\n\n _isShown(element = this._element) {\n return element.classList.contains(CLASS_NAME_SHOW)\n }\n\n // Private\n _configAfterMerge(config) {\n config.toggle = Boolean(config.toggle) // Coerce string values\n config.parent = getElement(config.parent)\n return config\n }\n\n _getDimension() {\n return this._element.classList.contains(CLASS_NAME_HORIZONTAL) ? WIDTH : HEIGHT\n }\n\n _initializeChildren() {\n if (!this._config.parent) {\n return\n }\n\n const children = this._getFirstLevelChildren(SELECTOR_DATA_TOGGLE)\n\n for (const element of children) {\n const selected = SelectorEngine.getElementFromSelector(element)\n\n if (selected) {\n this._addAriaAndCollapsedClass([element], this._isShown(selected))\n }\n }\n }\n\n _getFirstLevelChildren(selector) {\n const children = SelectorEngine.find(CLASS_NAME_DEEPER_CHILDREN, this._config.parent)\n // remove children if greater depth\n return SelectorEngine.find(selector, this._config.parent).filter(element => !children.includes(element))\n }\n\n _addAriaAndCollapsedClass(triggerArray, isOpen) {\n if (!triggerArray.length) {\n return\n }\n\n for (const element of triggerArray) {\n element.classList.toggle(CLASS_NAME_COLLAPSED, !isOpen)\n element.setAttribute('aria-expanded', isOpen)\n }\n }\n\n // Static\n static jQueryInterface(config) {\n const _config = {}\n if (typeof config === 'string' && /show|hide/.test(config)) {\n _config.toggle = false\n }\n\n return this.each(function () {\n const data = Collapse.getOrCreateInstance(this, _config)\n\n if (typeof config === 'string') {\n if (typeof data[config] === 'undefined') {\n throw new TypeError(`No method named \"${config}\"`)\n }\n\n data[config]()\n }\n })\n }\n}\n\n/**\n * Data API implementation\n */\n\nEventHandler.on(document, EVENT_CLICK_DATA_API, SELECTOR_DATA_TOGGLE, function (event) {\n // preventDefault only for elements (which change the URL) not inside the collapsible element\n if (event.target.tagName === 'A' || (event.delegateTarget && event.delegateTarget.tagName === 'A')) {\n event.preventDefault()\n }\n\n for (const element of SelectorEngine.getMultipleElementsFromSelector(this)) {\n Collapse.getOrCreateInstance(element, { toggle: false }).toggle()\n }\n})\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Collapse)\n\nexport default Collapse\n","export var top = 'top';\nexport var bottom = 'bottom';\nexport var right = 'right';\nexport var left = 'left';\nexport var auto = 'auto';\nexport var basePlacements = [top, bottom, right, left];\nexport var start = 'start';\nexport var end = 'end';\nexport var clippingParents = 'clippingParents';\nexport var viewport = 'viewport';\nexport var popper = 'popper';\nexport var reference = 'reference';\nexport var variationPlacements = /*#__PURE__*/basePlacements.reduce(function (acc, placement) {\n return acc.concat([placement + \"-\" + start, placement + \"-\" + end]);\n}, []);\nexport var placements = /*#__PURE__*/[].concat(basePlacements, [auto]).reduce(function (acc, placement) {\n return acc.concat([placement, placement + \"-\" + start, placement + \"-\" + end]);\n}, []); // modifiers that need to read the DOM\n\nexport var beforeRead = 'beforeRead';\nexport var read = 'read';\nexport var afterRead = 'afterRead'; // pure-logic modifiers\n\nexport var beforeMain = 'beforeMain';\nexport var main = 'main';\nexport var afterMain = 'afterMain'; // modifier with the purpose to write to the DOM (or write into a framework state)\n\nexport var beforeWrite = 'beforeWrite';\nexport var write = 'write';\nexport var afterWrite = 'afterWrite';\nexport var modifierPhases = [beforeRead, read, afterRead, beforeMain, main, afterMain, beforeWrite, write, afterWrite];","export default function getNodeName(element) {\n return element ? (element.nodeName || '').toLowerCase() : null;\n}","export default function getWindow(node) {\n if (node == null) {\n return window;\n }\n\n if (node.toString() !== '[object Window]') {\n var ownerDocument = node.ownerDocument;\n return ownerDocument ? ownerDocument.defaultView || window : window;\n }\n\n return node;\n}","import getWindow from \"./getWindow.js\";\n\nfunction isElement(node) {\n var OwnElement = getWindow(node).Element;\n return node instanceof OwnElement || node instanceof Element;\n}\n\nfunction isHTMLElement(node) {\n var OwnElement = getWindow(node).HTMLElement;\n return node instanceof OwnElement || node instanceof HTMLElement;\n}\n\nfunction isShadowRoot(node) {\n // IE 11 has no ShadowRoot\n if (typeof ShadowRoot === 'undefined') {\n return false;\n }\n\n var OwnElement = getWindow(node).ShadowRoot;\n return node instanceof OwnElement || node instanceof ShadowRoot;\n}\n\nexport { isElement, isHTMLElement, isShadowRoot };","import getNodeName from \"../dom-utils/getNodeName.js\";\nimport { isHTMLElement } from \"../dom-utils/instanceOf.js\"; // This modifier takes the styles prepared by the `computeStyles` modifier\n// and applies them to the HTMLElements such as popper and arrow\n\nfunction applyStyles(_ref) {\n var state = _ref.state;\n Object.keys(state.elements).forEach(function (name) {\n var style = state.styles[name] || {};\n var attributes = state.attributes[name] || {};\n var element = state.elements[name]; // arrow is optional + virtual elements\n\n if (!isHTMLElement(element) || !getNodeName(element)) {\n return;\n } // Flow doesn't support to extend this property, but it's the most\n // effective way to apply styles to an HTMLElement\n // $FlowFixMe[cannot-write]\n\n\n Object.assign(element.style, style);\n Object.keys(attributes).forEach(function (name) {\n var value = attributes[name];\n\n if (value === false) {\n element.removeAttribute(name);\n } else {\n element.setAttribute(name, value === true ? '' : value);\n }\n });\n });\n}\n\nfunction effect(_ref2) {\n var state = _ref2.state;\n var initialStyles = {\n popper: {\n position: state.options.strategy,\n left: '0',\n top: '0',\n margin: '0'\n },\n arrow: {\n position: 'absolute'\n },\n reference: {}\n };\n Object.assign(state.elements.popper.style, initialStyles.popper);\n state.styles = initialStyles;\n\n if (state.elements.arrow) {\n Object.assign(state.elements.arrow.style, initialStyles.arrow);\n }\n\n return function () {\n Object.keys(state.elements).forEach(function (name) {\n var element = state.elements[name];\n var attributes = state.attributes[name] || {};\n var styleProperties = Object.keys(state.styles.hasOwnProperty(name) ? state.styles[name] : initialStyles[name]); // Set all values to an empty string to unset them\n\n var style = styleProperties.reduce(function (style, property) {\n style[property] = '';\n return style;\n }, {}); // arrow is optional + virtual elements\n\n if (!isHTMLElement(element) || !getNodeName(element)) {\n return;\n }\n\n Object.assign(element.style, style);\n Object.keys(attributes).forEach(function (attribute) {\n element.removeAttribute(attribute);\n });\n });\n };\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'applyStyles',\n enabled: true,\n phase: 'write',\n fn: applyStyles,\n effect: effect,\n requires: ['computeStyles']\n};","import { auto } from \"../enums.js\";\nexport default function getBasePlacement(placement) {\n return placement.split('-')[0];\n}","export var max = Math.max;\nexport var min = Math.min;\nexport var round = Math.round;","export default function getUAString() {\n var uaData = navigator.userAgentData;\n\n if (uaData != null && uaData.brands && Array.isArray(uaData.brands)) {\n return uaData.brands.map(function (item) {\n return item.brand + \"/\" + item.version;\n }).join(' ');\n }\n\n return navigator.userAgent;\n}","import getUAString from \"../utils/userAgent.js\";\nexport default function isLayoutViewport() {\n return !/^((?!chrome|android).)*safari/i.test(getUAString());\n}","import { isElement, isHTMLElement } from \"./instanceOf.js\";\nimport { round } from \"../utils/math.js\";\nimport getWindow from \"./getWindow.js\";\nimport isLayoutViewport from \"./isLayoutViewport.js\";\nexport default function getBoundingClientRect(element, includeScale, isFixedStrategy) {\n if (includeScale === void 0) {\n includeScale = false;\n }\n\n if (isFixedStrategy === void 0) {\n isFixedStrategy = false;\n }\n\n var clientRect = element.getBoundingClientRect();\n var scaleX = 1;\n var scaleY = 1;\n\n if (includeScale && isHTMLElement(element)) {\n scaleX = element.offsetWidth > 0 ? round(clientRect.width) / element.offsetWidth || 1 : 1;\n scaleY = element.offsetHeight > 0 ? round(clientRect.height) / element.offsetHeight || 1 : 1;\n }\n\n var _ref = isElement(element) ? getWindow(element) : window,\n visualViewport = _ref.visualViewport;\n\n var addVisualOffsets = !isLayoutViewport() && isFixedStrategy;\n var x = (clientRect.left + (addVisualOffsets && visualViewport ? visualViewport.offsetLeft : 0)) / scaleX;\n var y = (clientRect.top + (addVisualOffsets && visualViewport ? visualViewport.offsetTop : 0)) / scaleY;\n var width = clientRect.width / scaleX;\n var height = clientRect.height / scaleY;\n return {\n width: width,\n height: height,\n top: y,\n right: x + width,\n bottom: y + height,\n left: x,\n x: x,\n y: y\n };\n}","import getBoundingClientRect from \"./getBoundingClientRect.js\"; // Returns the layout rect of an element relative to its offsetParent. Layout\n// means it doesn't take into account transforms.\n\nexport default function getLayoutRect(element) {\n var clientRect = getBoundingClientRect(element); // Use the clientRect sizes if it's not been transformed.\n // Fixes https://github.com/popperjs/popper-core/issues/1223\n\n var width = element.offsetWidth;\n var height = element.offsetHeight;\n\n if (Math.abs(clientRect.width - width) <= 1) {\n width = clientRect.width;\n }\n\n if (Math.abs(clientRect.height - height) <= 1) {\n height = clientRect.height;\n }\n\n return {\n x: element.offsetLeft,\n y: element.offsetTop,\n width: width,\n height: height\n };\n}","import { isShadowRoot } from \"./instanceOf.js\";\nexport default function contains(parent, child) {\n var rootNode = child.getRootNode && child.getRootNode(); // First, attempt with faster native method\n\n if (parent.contains(child)) {\n return true;\n } // then fallback to custom implementation with Shadow DOM support\n else if (rootNode && isShadowRoot(rootNode)) {\n var next = child;\n\n do {\n if (next && parent.isSameNode(next)) {\n return true;\n } // $FlowFixMe[prop-missing]: need a better way to handle this...\n\n\n next = next.parentNode || next.host;\n } while (next);\n } // Give up, the result is false\n\n\n return false;\n}","import getWindow from \"./getWindow.js\";\nexport default function getComputedStyle(element) {\n return getWindow(element).getComputedStyle(element);\n}","import getNodeName from \"./getNodeName.js\";\nexport default function isTableElement(element) {\n return ['table', 'td', 'th'].indexOf(getNodeName(element)) >= 0;\n}","import { isElement } from \"./instanceOf.js\";\nexport default function getDocumentElement(element) {\n // $FlowFixMe[incompatible-return]: assume body is always available\n return ((isElement(element) ? element.ownerDocument : // $FlowFixMe[prop-missing]\n element.document) || window.document).documentElement;\n}","import getNodeName from \"./getNodeName.js\";\nimport getDocumentElement from \"./getDocumentElement.js\";\nimport { isShadowRoot } from \"./instanceOf.js\";\nexport default function getParentNode(element) {\n if (getNodeName(element) === 'html') {\n return element;\n }\n\n return (// this is a quicker (but less type safe) way to save quite some bytes from the bundle\n // $FlowFixMe[incompatible-return]\n // $FlowFixMe[prop-missing]\n element.assignedSlot || // step into the shadow DOM of the parent of a slotted node\n element.parentNode || ( // DOM Element detected\n isShadowRoot(element) ? element.host : null) || // ShadowRoot detected\n // $FlowFixMe[incompatible-call]: HTMLElement is a Node\n getDocumentElement(element) // fallback\n\n );\n}","import getWindow from \"./getWindow.js\";\nimport getNodeName from \"./getNodeName.js\";\nimport getComputedStyle from \"./getComputedStyle.js\";\nimport { isHTMLElement, isShadowRoot } from \"./instanceOf.js\";\nimport isTableElement from \"./isTableElement.js\";\nimport getParentNode from \"./getParentNode.js\";\nimport getUAString from \"../utils/userAgent.js\";\n\nfunction getTrueOffsetParent(element) {\n if (!isHTMLElement(element) || // https://github.com/popperjs/popper-core/issues/837\n getComputedStyle(element).position === 'fixed') {\n return null;\n }\n\n return element.offsetParent;\n} // `.offsetParent` reports `null` for fixed elements, while absolute elements\n// return the containing block\n\n\nfunction getContainingBlock(element) {\n var isFirefox = /firefox/i.test(getUAString());\n var isIE = /Trident/i.test(getUAString());\n\n if (isIE && isHTMLElement(element)) {\n // In IE 9, 10 and 11 fixed elements containing block is always established by the viewport\n var elementCss = getComputedStyle(element);\n\n if (elementCss.position === 'fixed') {\n return null;\n }\n }\n\n var currentNode = getParentNode(element);\n\n if (isShadowRoot(currentNode)) {\n currentNode = currentNode.host;\n }\n\n while (isHTMLElement(currentNode) && ['html', 'body'].indexOf(getNodeName(currentNode)) < 0) {\n var css = getComputedStyle(currentNode); // This is non-exhaustive but covers the most common CSS properties that\n // create a containing block.\n // https://developer.mozilla.org/en-US/docs/Web/CSS/Containing_block#identifying_the_containing_block\n\n if (css.transform !== 'none' || css.perspective !== 'none' || css.contain === 'paint' || ['transform', 'perspective'].indexOf(css.willChange) !== -1 || isFirefox && css.willChange === 'filter' || isFirefox && css.filter && css.filter !== 'none') {\n return currentNode;\n } else {\n currentNode = currentNode.parentNode;\n }\n }\n\n return null;\n} // Gets the closest ancestor positioned element. Handles some edge cases,\n// such as table ancestors and cross browser bugs.\n\n\nexport default function getOffsetParent(element) {\n var window = getWindow(element);\n var offsetParent = getTrueOffsetParent(element);\n\n while (offsetParent && isTableElement(offsetParent) && getComputedStyle(offsetParent).position === 'static') {\n offsetParent = getTrueOffsetParent(offsetParent);\n }\n\n if (offsetParent && (getNodeName(offsetParent) === 'html' || getNodeName(offsetParent) === 'body' && getComputedStyle(offsetParent).position === 'static')) {\n return window;\n }\n\n return offsetParent || getContainingBlock(element) || window;\n}","export default function getMainAxisFromPlacement(placement) {\n return ['top', 'bottom'].indexOf(placement) >= 0 ? 'x' : 'y';\n}","import { max as mathMax, min as mathMin } from \"./math.js\";\nexport function within(min, value, max) {\n return mathMax(min, mathMin(value, max));\n}\nexport function withinMaxClamp(min, value, max) {\n var v = within(min, value, max);\n return v > max ? max : v;\n}","import getFreshSideObject from \"./getFreshSideObject.js\";\nexport default function mergePaddingObject(paddingObject) {\n return Object.assign({}, getFreshSideObject(), paddingObject);\n}","export default function getFreshSideObject() {\n return {\n top: 0,\n right: 0,\n bottom: 0,\n left: 0\n };\n}","export default function expandToHashMap(value, keys) {\n return keys.reduce(function (hashMap, key) {\n hashMap[key] = value;\n return hashMap;\n }, {});\n}","import getBasePlacement from \"../utils/getBasePlacement.js\";\nimport getLayoutRect from \"../dom-utils/getLayoutRect.js\";\nimport contains from \"../dom-utils/contains.js\";\nimport getOffsetParent from \"../dom-utils/getOffsetParent.js\";\nimport getMainAxisFromPlacement from \"../utils/getMainAxisFromPlacement.js\";\nimport { within } from \"../utils/within.js\";\nimport mergePaddingObject from \"../utils/mergePaddingObject.js\";\nimport expandToHashMap from \"../utils/expandToHashMap.js\";\nimport { left, right, basePlacements, top, bottom } from \"../enums.js\"; // eslint-disable-next-line import/no-unused-modules\n\nvar toPaddingObject = function toPaddingObject(padding, state) {\n padding = typeof padding === 'function' ? padding(Object.assign({}, state.rects, {\n placement: state.placement\n })) : padding;\n return mergePaddingObject(typeof padding !== 'number' ? padding : expandToHashMap(padding, basePlacements));\n};\n\nfunction arrow(_ref) {\n var _state$modifiersData$;\n\n var state = _ref.state,\n name = _ref.name,\n options = _ref.options;\n var arrowElement = state.elements.arrow;\n var popperOffsets = state.modifiersData.popperOffsets;\n var basePlacement = getBasePlacement(state.placement);\n var axis = getMainAxisFromPlacement(basePlacement);\n var isVertical = [left, right].indexOf(basePlacement) >= 0;\n var len = isVertical ? 'height' : 'width';\n\n if (!arrowElement || !popperOffsets) {\n return;\n }\n\n var paddingObject = toPaddingObject(options.padding, state);\n var arrowRect = getLayoutRect(arrowElement);\n var minProp = axis === 'y' ? top : left;\n var maxProp = axis === 'y' ? bottom : right;\n var endDiff = state.rects.reference[len] + state.rects.reference[axis] - popperOffsets[axis] - state.rects.popper[len];\n var startDiff = popperOffsets[axis] - state.rects.reference[axis];\n var arrowOffsetParent = getOffsetParent(arrowElement);\n var clientSize = arrowOffsetParent ? axis === 'y' ? arrowOffsetParent.clientHeight || 0 : arrowOffsetParent.clientWidth || 0 : 0;\n var centerToReference = endDiff / 2 - startDiff / 2; // Make sure the arrow doesn't overflow the popper if the center point is\n // outside of the popper bounds\n\n var min = paddingObject[minProp];\n var max = clientSize - arrowRect[len] - paddingObject[maxProp];\n var center = clientSize / 2 - arrowRect[len] / 2 + centerToReference;\n var offset = within(min, center, max); // Prevents breaking syntax highlighting...\n\n var axisProp = axis;\n state.modifiersData[name] = (_state$modifiersData$ = {}, _state$modifiersData$[axisProp] = offset, _state$modifiersData$.centerOffset = offset - center, _state$modifiersData$);\n}\n\nfunction effect(_ref2) {\n var state = _ref2.state,\n options = _ref2.options;\n var _options$element = options.element,\n arrowElement = _options$element === void 0 ? '[data-popper-arrow]' : _options$element;\n\n if (arrowElement == null) {\n return;\n } // CSS selector\n\n\n if (typeof arrowElement === 'string') {\n arrowElement = state.elements.popper.querySelector(arrowElement);\n\n if (!arrowElement) {\n return;\n }\n }\n\n if (!contains(state.elements.popper, arrowElement)) {\n return;\n }\n\n state.elements.arrow = arrowElement;\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'arrow',\n enabled: true,\n phase: 'main',\n fn: arrow,\n effect: effect,\n requires: ['popperOffsets'],\n requiresIfExists: ['preventOverflow']\n};","export default function getVariation(placement) {\n return placement.split('-')[1];\n}","import { top, left, right, bottom, end } from \"../enums.js\";\nimport getOffsetParent from \"../dom-utils/getOffsetParent.js\";\nimport getWindow from \"../dom-utils/getWindow.js\";\nimport getDocumentElement from \"../dom-utils/getDocumentElement.js\";\nimport getComputedStyle from \"../dom-utils/getComputedStyle.js\";\nimport getBasePlacement from \"../utils/getBasePlacement.js\";\nimport getVariation from \"../utils/getVariation.js\";\nimport { round } from \"../utils/math.js\"; // eslint-disable-next-line import/no-unused-modules\n\nvar unsetSides = {\n top: 'auto',\n right: 'auto',\n bottom: 'auto',\n left: 'auto'\n}; // Round the offsets to the nearest suitable subpixel based on the DPR.\n// Zooming can change the DPR, but it seems to report a value that will\n// cleanly divide the values into the appropriate subpixels.\n\nfunction roundOffsetsByDPR(_ref, win) {\n var x = _ref.x,\n y = _ref.y;\n var dpr = win.devicePixelRatio || 1;\n return {\n x: round(x * dpr) / dpr || 0,\n y: round(y * dpr) / dpr || 0\n };\n}\n\nexport function mapToStyles(_ref2) {\n var _Object$assign2;\n\n var popper = _ref2.popper,\n popperRect = _ref2.popperRect,\n placement = _ref2.placement,\n variation = _ref2.variation,\n offsets = _ref2.offsets,\n position = _ref2.position,\n gpuAcceleration = _ref2.gpuAcceleration,\n adaptive = _ref2.adaptive,\n roundOffsets = _ref2.roundOffsets,\n isFixed = _ref2.isFixed;\n var _offsets$x = offsets.x,\n x = _offsets$x === void 0 ? 0 : _offsets$x,\n _offsets$y = offsets.y,\n y = _offsets$y === void 0 ? 0 : _offsets$y;\n\n var _ref3 = typeof roundOffsets === 'function' ? roundOffsets({\n x: x,\n y: y\n }) : {\n x: x,\n y: y\n };\n\n x = _ref3.x;\n y = _ref3.y;\n var hasX = offsets.hasOwnProperty('x');\n var hasY = offsets.hasOwnProperty('y');\n var sideX = left;\n var sideY = top;\n var win = window;\n\n if (adaptive) {\n var offsetParent = getOffsetParent(popper);\n var heightProp = 'clientHeight';\n var widthProp = 'clientWidth';\n\n if (offsetParent === getWindow(popper)) {\n offsetParent = getDocumentElement(popper);\n\n if (getComputedStyle(offsetParent).position !== 'static' && position === 'absolute') {\n heightProp = 'scrollHeight';\n widthProp = 'scrollWidth';\n }\n } // $FlowFixMe[incompatible-cast]: force type refinement, we compare offsetParent with window above, but Flow doesn't detect it\n\n\n offsetParent = offsetParent;\n\n if (placement === top || (placement === left || placement === right) && variation === end) {\n sideY = bottom;\n var offsetY = isFixed && offsetParent === win && win.visualViewport ? win.visualViewport.height : // $FlowFixMe[prop-missing]\n offsetParent[heightProp];\n y -= offsetY - popperRect.height;\n y *= gpuAcceleration ? 1 : -1;\n }\n\n if (placement === left || (placement === top || placement === bottom) && variation === end) {\n sideX = right;\n var offsetX = isFixed && offsetParent === win && win.visualViewport ? win.visualViewport.width : // $FlowFixMe[prop-missing]\n offsetParent[widthProp];\n x -= offsetX - popperRect.width;\n x *= gpuAcceleration ? 1 : -1;\n }\n }\n\n var commonStyles = Object.assign({\n position: position\n }, adaptive && unsetSides);\n\n var _ref4 = roundOffsets === true ? roundOffsetsByDPR({\n x: x,\n y: y\n }, getWindow(popper)) : {\n x: x,\n y: y\n };\n\n x = _ref4.x;\n y = _ref4.y;\n\n if (gpuAcceleration) {\n var _Object$assign;\n\n return Object.assign({}, commonStyles, (_Object$assign = {}, _Object$assign[sideY] = hasY ? '0' : '', _Object$assign[sideX] = hasX ? '0' : '', _Object$assign.transform = (win.devicePixelRatio || 1) <= 1 ? \"translate(\" + x + \"px, \" + y + \"px)\" : \"translate3d(\" + x + \"px, \" + y + \"px, 0)\", _Object$assign));\n }\n\n return Object.assign({}, commonStyles, (_Object$assign2 = {}, _Object$assign2[sideY] = hasY ? y + \"px\" : '', _Object$assign2[sideX] = hasX ? x + \"px\" : '', _Object$assign2.transform = '', _Object$assign2));\n}\n\nfunction computeStyles(_ref5) {\n var state = _ref5.state,\n options = _ref5.options;\n var _options$gpuAccelerat = options.gpuAcceleration,\n gpuAcceleration = _options$gpuAccelerat === void 0 ? true : _options$gpuAccelerat,\n _options$adaptive = options.adaptive,\n adaptive = _options$adaptive === void 0 ? true : _options$adaptive,\n _options$roundOffsets = options.roundOffsets,\n roundOffsets = _options$roundOffsets === void 0 ? true : _options$roundOffsets;\n var commonStyles = {\n placement: getBasePlacement(state.placement),\n variation: getVariation(state.placement),\n popper: state.elements.popper,\n popperRect: state.rects.popper,\n gpuAcceleration: gpuAcceleration,\n isFixed: state.options.strategy === 'fixed'\n };\n\n if (state.modifiersData.popperOffsets != null) {\n state.styles.popper = Object.assign({}, state.styles.popper, mapToStyles(Object.assign({}, commonStyles, {\n offsets: state.modifiersData.popperOffsets,\n position: state.options.strategy,\n adaptive: adaptive,\n roundOffsets: roundOffsets\n })));\n }\n\n if (state.modifiersData.arrow != null) {\n state.styles.arrow = Object.assign({}, state.styles.arrow, mapToStyles(Object.assign({}, commonStyles, {\n offsets: state.modifiersData.arrow,\n position: 'absolute',\n adaptive: false,\n roundOffsets: roundOffsets\n })));\n }\n\n state.attributes.popper = Object.assign({}, state.attributes.popper, {\n 'data-popper-placement': state.placement\n });\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'computeStyles',\n enabled: true,\n phase: 'beforeWrite',\n fn: computeStyles,\n data: {}\n};","import getWindow from \"../dom-utils/getWindow.js\"; // eslint-disable-next-line import/no-unused-modules\n\nvar passive = {\n passive: true\n};\n\nfunction effect(_ref) {\n var state = _ref.state,\n instance = _ref.instance,\n options = _ref.options;\n var _options$scroll = options.scroll,\n scroll = _options$scroll === void 0 ? true : _options$scroll,\n _options$resize = options.resize,\n resize = _options$resize === void 0 ? true : _options$resize;\n var window = getWindow(state.elements.popper);\n var scrollParents = [].concat(state.scrollParents.reference, state.scrollParents.popper);\n\n if (scroll) {\n scrollParents.forEach(function (scrollParent) {\n scrollParent.addEventListener('scroll', instance.update, passive);\n });\n }\n\n if (resize) {\n window.addEventListener('resize', instance.update, passive);\n }\n\n return function () {\n if (scroll) {\n scrollParents.forEach(function (scrollParent) {\n scrollParent.removeEventListener('scroll', instance.update, passive);\n });\n }\n\n if (resize) {\n window.removeEventListener('resize', instance.update, passive);\n }\n };\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'eventListeners',\n enabled: true,\n phase: 'write',\n fn: function fn() {},\n effect: effect,\n data: {}\n};","var hash = {\n left: 'right',\n right: 'left',\n bottom: 'top',\n top: 'bottom'\n};\nexport default function getOppositePlacement(placement) {\n return placement.replace(/left|right|bottom|top/g, function (matched) {\n return hash[matched];\n });\n}","var hash = {\n start: 'end',\n end: 'start'\n};\nexport default function getOppositeVariationPlacement(placement) {\n return placement.replace(/start|end/g, function (matched) {\n return hash[matched];\n });\n}","import getWindow from \"./getWindow.js\";\nexport default function getWindowScroll(node) {\n var win = getWindow(node);\n var scrollLeft = win.pageXOffset;\n var scrollTop = win.pageYOffset;\n return {\n scrollLeft: scrollLeft,\n scrollTop: scrollTop\n };\n}","import getBoundingClientRect from \"./getBoundingClientRect.js\";\nimport getDocumentElement from \"./getDocumentElement.js\";\nimport getWindowScroll from \"./getWindowScroll.js\";\nexport default function getWindowScrollBarX(element) {\n // If has a CSS width greater than the viewport, then this will be\n // incorrect for RTL.\n // Popper 1 is broken in this case and never had a bug report so let's assume\n // it's not an issue. I don't think anyone ever specifies width on \n // anyway.\n // Browsers where the left scrollbar doesn't cause an issue report `0` for\n // this (e.g. Edge 2019, IE11, Safari)\n return getBoundingClientRect(getDocumentElement(element)).left + getWindowScroll(element).scrollLeft;\n}","import getComputedStyle from \"./getComputedStyle.js\";\nexport default function isScrollParent(element) {\n // Firefox wants us to check `-x` and `-y` variations as well\n var _getComputedStyle = getComputedStyle(element),\n overflow = _getComputedStyle.overflow,\n overflowX = _getComputedStyle.overflowX,\n overflowY = _getComputedStyle.overflowY;\n\n return /auto|scroll|overlay|hidden/.test(overflow + overflowY + overflowX);\n}","import getParentNode from \"./getParentNode.js\";\nimport isScrollParent from \"./isScrollParent.js\";\nimport getNodeName from \"./getNodeName.js\";\nimport { isHTMLElement } from \"./instanceOf.js\";\nexport default function getScrollParent(node) {\n if (['html', 'body', '#document'].indexOf(getNodeName(node)) >= 0) {\n // $FlowFixMe[incompatible-return]: assume body is always available\n return node.ownerDocument.body;\n }\n\n if (isHTMLElement(node) && isScrollParent(node)) {\n return node;\n }\n\n return getScrollParent(getParentNode(node));\n}","import getScrollParent from \"./getScrollParent.js\";\nimport getParentNode from \"./getParentNode.js\";\nimport getWindow from \"./getWindow.js\";\nimport isScrollParent from \"./isScrollParent.js\";\n/*\ngiven a DOM element, return the list of all scroll parents, up the list of ancesors\nuntil we get to the top window object. This list is what we attach scroll listeners\nto, because if any of these parent elements scroll, we'll need to re-calculate the\nreference element's position.\n*/\n\nexport default function listScrollParents(element, list) {\n var _element$ownerDocumen;\n\n if (list === void 0) {\n list = [];\n }\n\n var scrollParent = getScrollParent(element);\n var isBody = scrollParent === ((_element$ownerDocumen = element.ownerDocument) == null ? void 0 : _element$ownerDocumen.body);\n var win = getWindow(scrollParent);\n var target = isBody ? [win].concat(win.visualViewport || [], isScrollParent(scrollParent) ? scrollParent : []) : scrollParent;\n var updatedList = list.concat(target);\n return isBody ? updatedList : // $FlowFixMe[incompatible-call]: isBody tells us target will be an HTMLElement here\n updatedList.concat(listScrollParents(getParentNode(target)));\n}","export default function rectToClientRect(rect) {\n return Object.assign({}, rect, {\n left: rect.x,\n top: rect.y,\n right: rect.x + rect.width,\n bottom: rect.y + rect.height\n });\n}","import { viewport } from \"../enums.js\";\nimport getViewportRect from \"./getViewportRect.js\";\nimport getDocumentRect from \"./getDocumentRect.js\";\nimport listScrollParents from \"./listScrollParents.js\";\nimport getOffsetParent from \"./getOffsetParent.js\";\nimport getDocumentElement from \"./getDocumentElement.js\";\nimport getComputedStyle from \"./getComputedStyle.js\";\nimport { isElement, isHTMLElement } from \"./instanceOf.js\";\nimport getBoundingClientRect from \"./getBoundingClientRect.js\";\nimport getParentNode from \"./getParentNode.js\";\nimport contains from \"./contains.js\";\nimport getNodeName from \"./getNodeName.js\";\nimport rectToClientRect from \"../utils/rectToClientRect.js\";\nimport { max, min } from \"../utils/math.js\";\n\nfunction getInnerBoundingClientRect(element, strategy) {\n var rect = getBoundingClientRect(element, false, strategy === 'fixed');\n rect.top = rect.top + element.clientTop;\n rect.left = rect.left + element.clientLeft;\n rect.bottom = rect.top + element.clientHeight;\n rect.right = rect.left + element.clientWidth;\n rect.width = element.clientWidth;\n rect.height = element.clientHeight;\n rect.x = rect.left;\n rect.y = rect.top;\n return rect;\n}\n\nfunction getClientRectFromMixedType(element, clippingParent, strategy) {\n return clippingParent === viewport ? rectToClientRect(getViewportRect(element, strategy)) : isElement(clippingParent) ? getInnerBoundingClientRect(clippingParent, strategy) : rectToClientRect(getDocumentRect(getDocumentElement(element)));\n} // A \"clipping parent\" is an overflowable container with the characteristic of\n// clipping (or hiding) overflowing elements with a position different from\n// `initial`\n\n\nfunction getClippingParents(element) {\n var clippingParents = listScrollParents(getParentNode(element));\n var canEscapeClipping = ['absolute', 'fixed'].indexOf(getComputedStyle(element).position) >= 0;\n var clipperElement = canEscapeClipping && isHTMLElement(element) ? getOffsetParent(element) : element;\n\n if (!isElement(clipperElement)) {\n return [];\n } // $FlowFixMe[incompatible-return]: https://github.com/facebook/flow/issues/1414\n\n\n return clippingParents.filter(function (clippingParent) {\n return isElement(clippingParent) && contains(clippingParent, clipperElement) && getNodeName(clippingParent) !== 'body';\n });\n} // Gets the maximum area that the element is visible in due to any number of\n// clipping parents\n\n\nexport default function getClippingRect(element, boundary, rootBoundary, strategy) {\n var mainClippingParents = boundary === 'clippingParents' ? getClippingParents(element) : [].concat(boundary);\n var clippingParents = [].concat(mainClippingParents, [rootBoundary]);\n var firstClippingParent = clippingParents[0];\n var clippingRect = clippingParents.reduce(function (accRect, clippingParent) {\n var rect = getClientRectFromMixedType(element, clippingParent, strategy);\n accRect.top = max(rect.top, accRect.top);\n accRect.right = min(rect.right, accRect.right);\n accRect.bottom = min(rect.bottom, accRect.bottom);\n accRect.left = max(rect.left, accRect.left);\n return accRect;\n }, getClientRectFromMixedType(element, firstClippingParent, strategy));\n clippingRect.width = clippingRect.right - clippingRect.left;\n clippingRect.height = clippingRect.bottom - clippingRect.top;\n clippingRect.x = clippingRect.left;\n clippingRect.y = clippingRect.top;\n return clippingRect;\n}","import getWindow from \"./getWindow.js\";\nimport getDocumentElement from \"./getDocumentElement.js\";\nimport getWindowScrollBarX from \"./getWindowScrollBarX.js\";\nimport isLayoutViewport from \"./isLayoutViewport.js\";\nexport default function getViewportRect(element, strategy) {\n var win = getWindow(element);\n var html = getDocumentElement(element);\n var visualViewport = win.visualViewport;\n var width = html.clientWidth;\n var height = html.clientHeight;\n var x = 0;\n var y = 0;\n\n if (visualViewport) {\n width = visualViewport.width;\n height = visualViewport.height;\n var layoutViewport = isLayoutViewport();\n\n if (layoutViewport || !layoutViewport && strategy === 'fixed') {\n x = visualViewport.offsetLeft;\n y = visualViewport.offsetTop;\n }\n }\n\n return {\n width: width,\n height: height,\n x: x + getWindowScrollBarX(element),\n y: y\n };\n}","import getDocumentElement from \"./getDocumentElement.js\";\nimport getComputedStyle from \"./getComputedStyle.js\";\nimport getWindowScrollBarX from \"./getWindowScrollBarX.js\";\nimport getWindowScroll from \"./getWindowScroll.js\";\nimport { max } from \"../utils/math.js\"; // Gets the entire size of the scrollable document area, even extending outside\n// of the `` and `` rect bounds if horizontally scrollable\n\nexport default function getDocumentRect(element) {\n var _element$ownerDocumen;\n\n var html = getDocumentElement(element);\n var winScroll = getWindowScroll(element);\n var body = (_element$ownerDocumen = element.ownerDocument) == null ? void 0 : _element$ownerDocumen.body;\n var width = max(html.scrollWidth, html.clientWidth, body ? body.scrollWidth : 0, body ? body.clientWidth : 0);\n var height = max(html.scrollHeight, html.clientHeight, body ? body.scrollHeight : 0, body ? body.clientHeight : 0);\n var x = -winScroll.scrollLeft + getWindowScrollBarX(element);\n var y = -winScroll.scrollTop;\n\n if (getComputedStyle(body || html).direction === 'rtl') {\n x += max(html.clientWidth, body ? body.clientWidth : 0) - width;\n }\n\n return {\n width: width,\n height: height,\n x: x,\n y: y\n };\n}","import getBasePlacement from \"./getBasePlacement.js\";\nimport getVariation from \"./getVariation.js\";\nimport getMainAxisFromPlacement from \"./getMainAxisFromPlacement.js\";\nimport { top, right, bottom, left, start, end } from \"../enums.js\";\nexport default function computeOffsets(_ref) {\n var reference = _ref.reference,\n element = _ref.element,\n placement = _ref.placement;\n var basePlacement = placement ? getBasePlacement(placement) : null;\n var variation = placement ? getVariation(placement) : null;\n var commonX = reference.x + reference.width / 2 - element.width / 2;\n var commonY = reference.y + reference.height / 2 - element.height / 2;\n var offsets;\n\n switch (basePlacement) {\n case top:\n offsets = {\n x: commonX,\n y: reference.y - element.height\n };\n break;\n\n case bottom:\n offsets = {\n x: commonX,\n y: reference.y + reference.height\n };\n break;\n\n case right:\n offsets = {\n x: reference.x + reference.width,\n y: commonY\n };\n break;\n\n case left:\n offsets = {\n x: reference.x - element.width,\n y: commonY\n };\n break;\n\n default:\n offsets = {\n x: reference.x,\n y: reference.y\n };\n }\n\n var mainAxis = basePlacement ? getMainAxisFromPlacement(basePlacement) : null;\n\n if (mainAxis != null) {\n var len = mainAxis === 'y' ? 'height' : 'width';\n\n switch (variation) {\n case start:\n offsets[mainAxis] = offsets[mainAxis] - (reference[len] / 2 - element[len] / 2);\n break;\n\n case end:\n offsets[mainAxis] = offsets[mainAxis] + (reference[len] / 2 - element[len] / 2);\n break;\n\n default:\n }\n }\n\n return offsets;\n}","import getClippingRect from \"../dom-utils/getClippingRect.js\";\nimport getDocumentElement from \"../dom-utils/getDocumentElement.js\";\nimport getBoundingClientRect from \"../dom-utils/getBoundingClientRect.js\";\nimport computeOffsets from \"./computeOffsets.js\";\nimport rectToClientRect from \"./rectToClientRect.js\";\nimport { clippingParents, reference, popper, bottom, top, right, basePlacements, viewport } from \"../enums.js\";\nimport { isElement } from \"../dom-utils/instanceOf.js\";\nimport mergePaddingObject from \"./mergePaddingObject.js\";\nimport expandToHashMap from \"./expandToHashMap.js\"; // eslint-disable-next-line import/no-unused-modules\n\nexport default function detectOverflow(state, options) {\n if (options === void 0) {\n options = {};\n }\n\n var _options = options,\n _options$placement = _options.placement,\n placement = _options$placement === void 0 ? state.placement : _options$placement,\n _options$strategy = _options.strategy,\n strategy = _options$strategy === void 0 ? state.strategy : _options$strategy,\n _options$boundary = _options.boundary,\n boundary = _options$boundary === void 0 ? clippingParents : _options$boundary,\n _options$rootBoundary = _options.rootBoundary,\n rootBoundary = _options$rootBoundary === void 0 ? viewport : _options$rootBoundary,\n _options$elementConte = _options.elementContext,\n elementContext = _options$elementConte === void 0 ? popper : _options$elementConte,\n _options$altBoundary = _options.altBoundary,\n altBoundary = _options$altBoundary === void 0 ? false : _options$altBoundary,\n _options$padding = _options.padding,\n padding = _options$padding === void 0 ? 0 : _options$padding;\n var paddingObject = mergePaddingObject(typeof padding !== 'number' ? padding : expandToHashMap(padding, basePlacements));\n var altContext = elementContext === popper ? reference : popper;\n var popperRect = state.rects.popper;\n var element = state.elements[altBoundary ? altContext : elementContext];\n var clippingClientRect = getClippingRect(isElement(element) ? element : element.contextElement || getDocumentElement(state.elements.popper), boundary, rootBoundary, strategy);\n var referenceClientRect = getBoundingClientRect(state.elements.reference);\n var popperOffsets = computeOffsets({\n reference: referenceClientRect,\n element: popperRect,\n strategy: 'absolute',\n placement: placement\n });\n var popperClientRect = rectToClientRect(Object.assign({}, popperRect, popperOffsets));\n var elementClientRect = elementContext === popper ? popperClientRect : referenceClientRect; // positive = overflowing the clipping rect\n // 0 or negative = within the clipping rect\n\n var overflowOffsets = {\n top: clippingClientRect.top - elementClientRect.top + paddingObject.top,\n bottom: elementClientRect.bottom - clippingClientRect.bottom + paddingObject.bottom,\n left: clippingClientRect.left - elementClientRect.left + paddingObject.left,\n right: elementClientRect.right - clippingClientRect.right + paddingObject.right\n };\n var offsetData = state.modifiersData.offset; // Offsets can be applied only to the popper element\n\n if (elementContext === popper && offsetData) {\n var offset = offsetData[placement];\n Object.keys(overflowOffsets).forEach(function (key) {\n var multiply = [right, bottom].indexOf(key) >= 0 ? 1 : -1;\n var axis = [top, bottom].indexOf(key) >= 0 ? 'y' : 'x';\n overflowOffsets[key] += offset[axis] * multiply;\n });\n }\n\n return overflowOffsets;\n}","import getVariation from \"./getVariation.js\";\nimport { variationPlacements, basePlacements, placements as allPlacements } from \"../enums.js\";\nimport detectOverflow from \"./detectOverflow.js\";\nimport getBasePlacement from \"./getBasePlacement.js\";\nexport default function computeAutoPlacement(state, options) {\n if (options === void 0) {\n options = {};\n }\n\n var _options = options,\n placement = _options.placement,\n boundary = _options.boundary,\n rootBoundary = _options.rootBoundary,\n padding = _options.padding,\n flipVariations = _options.flipVariations,\n _options$allowedAutoP = _options.allowedAutoPlacements,\n allowedAutoPlacements = _options$allowedAutoP === void 0 ? allPlacements : _options$allowedAutoP;\n var variation = getVariation(placement);\n var placements = variation ? flipVariations ? variationPlacements : variationPlacements.filter(function (placement) {\n return getVariation(placement) === variation;\n }) : basePlacements;\n var allowedPlacements = placements.filter(function (placement) {\n return allowedAutoPlacements.indexOf(placement) >= 0;\n });\n\n if (allowedPlacements.length === 0) {\n allowedPlacements = placements;\n } // $FlowFixMe[incompatible-type]: Flow seems to have problems with two array unions...\n\n\n var overflows = allowedPlacements.reduce(function (acc, placement) {\n acc[placement] = detectOverflow(state, {\n placement: placement,\n boundary: boundary,\n rootBoundary: rootBoundary,\n padding: padding\n })[getBasePlacement(placement)];\n return acc;\n }, {});\n return Object.keys(overflows).sort(function (a, b) {\n return overflows[a] - overflows[b];\n });\n}","import getOppositePlacement from \"../utils/getOppositePlacement.js\";\nimport getBasePlacement from \"../utils/getBasePlacement.js\";\nimport getOppositeVariationPlacement from \"../utils/getOppositeVariationPlacement.js\";\nimport detectOverflow from \"../utils/detectOverflow.js\";\nimport computeAutoPlacement from \"../utils/computeAutoPlacement.js\";\nimport { bottom, top, start, right, left, auto } from \"../enums.js\";\nimport getVariation from \"../utils/getVariation.js\"; // eslint-disable-next-line import/no-unused-modules\n\nfunction getExpandedFallbackPlacements(placement) {\n if (getBasePlacement(placement) === auto) {\n return [];\n }\n\n var oppositePlacement = getOppositePlacement(placement);\n return [getOppositeVariationPlacement(placement), oppositePlacement, getOppositeVariationPlacement(oppositePlacement)];\n}\n\nfunction flip(_ref) {\n var state = _ref.state,\n options = _ref.options,\n name = _ref.name;\n\n if (state.modifiersData[name]._skip) {\n return;\n }\n\n var _options$mainAxis = options.mainAxis,\n checkMainAxis = _options$mainAxis === void 0 ? true : _options$mainAxis,\n _options$altAxis = options.altAxis,\n checkAltAxis = _options$altAxis === void 0 ? true : _options$altAxis,\n specifiedFallbackPlacements = options.fallbackPlacements,\n padding = options.padding,\n boundary = options.boundary,\n rootBoundary = options.rootBoundary,\n altBoundary = options.altBoundary,\n _options$flipVariatio = options.flipVariations,\n flipVariations = _options$flipVariatio === void 0 ? true : _options$flipVariatio,\n allowedAutoPlacements = options.allowedAutoPlacements;\n var preferredPlacement = state.options.placement;\n var basePlacement = getBasePlacement(preferredPlacement);\n var isBasePlacement = basePlacement === preferredPlacement;\n var fallbackPlacements = specifiedFallbackPlacements || (isBasePlacement || !flipVariations ? [getOppositePlacement(preferredPlacement)] : getExpandedFallbackPlacements(preferredPlacement));\n var placements = [preferredPlacement].concat(fallbackPlacements).reduce(function (acc, placement) {\n return acc.concat(getBasePlacement(placement) === auto ? computeAutoPlacement(state, {\n placement: placement,\n boundary: boundary,\n rootBoundary: rootBoundary,\n padding: padding,\n flipVariations: flipVariations,\n allowedAutoPlacements: allowedAutoPlacements\n }) : placement);\n }, []);\n var referenceRect = state.rects.reference;\n var popperRect = state.rects.popper;\n var checksMap = new Map();\n var makeFallbackChecks = true;\n var firstFittingPlacement = placements[0];\n\n for (var i = 0; i < placements.length; i++) {\n var placement = placements[i];\n\n var _basePlacement = getBasePlacement(placement);\n\n var isStartVariation = getVariation(placement) === start;\n var isVertical = [top, bottom].indexOf(_basePlacement) >= 0;\n var len = isVertical ? 'width' : 'height';\n var overflow = detectOverflow(state, {\n placement: placement,\n boundary: boundary,\n rootBoundary: rootBoundary,\n altBoundary: altBoundary,\n padding: padding\n });\n var mainVariationSide = isVertical ? isStartVariation ? right : left : isStartVariation ? bottom : top;\n\n if (referenceRect[len] > popperRect[len]) {\n mainVariationSide = getOppositePlacement(mainVariationSide);\n }\n\n var altVariationSide = getOppositePlacement(mainVariationSide);\n var checks = [];\n\n if (checkMainAxis) {\n checks.push(overflow[_basePlacement] <= 0);\n }\n\n if (checkAltAxis) {\n checks.push(overflow[mainVariationSide] <= 0, overflow[altVariationSide] <= 0);\n }\n\n if (checks.every(function (check) {\n return check;\n })) {\n firstFittingPlacement = placement;\n makeFallbackChecks = false;\n break;\n }\n\n checksMap.set(placement, checks);\n }\n\n if (makeFallbackChecks) {\n // `2` may be desired in some cases – research later\n var numberOfChecks = flipVariations ? 3 : 1;\n\n var _loop = function _loop(_i) {\n var fittingPlacement = placements.find(function (placement) {\n var checks = checksMap.get(placement);\n\n if (checks) {\n return checks.slice(0, _i).every(function (check) {\n return check;\n });\n }\n });\n\n if (fittingPlacement) {\n firstFittingPlacement = fittingPlacement;\n return \"break\";\n }\n };\n\n for (var _i = numberOfChecks; _i > 0; _i--) {\n var _ret = _loop(_i);\n\n if (_ret === \"break\") break;\n }\n }\n\n if (state.placement !== firstFittingPlacement) {\n state.modifiersData[name]._skip = true;\n state.placement = firstFittingPlacement;\n state.reset = true;\n }\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'flip',\n enabled: true,\n phase: 'main',\n fn: flip,\n requiresIfExists: ['offset'],\n data: {\n _skip: false\n }\n};","import { top, bottom, left, right } from \"../enums.js\";\nimport detectOverflow from \"../utils/detectOverflow.js\";\n\nfunction getSideOffsets(overflow, rect, preventedOffsets) {\n if (preventedOffsets === void 0) {\n preventedOffsets = {\n x: 0,\n y: 0\n };\n }\n\n return {\n top: overflow.top - rect.height - preventedOffsets.y,\n right: overflow.right - rect.width + preventedOffsets.x,\n bottom: overflow.bottom - rect.height + preventedOffsets.y,\n left: overflow.left - rect.width - preventedOffsets.x\n };\n}\n\nfunction isAnySideFullyClipped(overflow) {\n return [top, right, bottom, left].some(function (side) {\n return overflow[side] >= 0;\n });\n}\n\nfunction hide(_ref) {\n var state = _ref.state,\n name = _ref.name;\n var referenceRect = state.rects.reference;\n var popperRect = state.rects.popper;\n var preventedOffsets = state.modifiersData.preventOverflow;\n var referenceOverflow = detectOverflow(state, {\n elementContext: 'reference'\n });\n var popperAltOverflow = detectOverflow(state, {\n altBoundary: true\n });\n var referenceClippingOffsets = getSideOffsets(referenceOverflow, referenceRect);\n var popperEscapeOffsets = getSideOffsets(popperAltOverflow, popperRect, preventedOffsets);\n var isReferenceHidden = isAnySideFullyClipped(referenceClippingOffsets);\n var hasPopperEscaped = isAnySideFullyClipped(popperEscapeOffsets);\n state.modifiersData[name] = {\n referenceClippingOffsets: referenceClippingOffsets,\n popperEscapeOffsets: popperEscapeOffsets,\n isReferenceHidden: isReferenceHidden,\n hasPopperEscaped: hasPopperEscaped\n };\n state.attributes.popper = Object.assign({}, state.attributes.popper, {\n 'data-popper-reference-hidden': isReferenceHidden,\n 'data-popper-escaped': hasPopperEscaped\n });\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'hide',\n enabled: true,\n phase: 'main',\n requiresIfExists: ['preventOverflow'],\n fn: hide\n};","import getBasePlacement from \"../utils/getBasePlacement.js\";\nimport { top, left, right, placements } from \"../enums.js\"; // eslint-disable-next-line import/no-unused-modules\n\nexport function distanceAndSkiddingToXY(placement, rects, offset) {\n var basePlacement = getBasePlacement(placement);\n var invertDistance = [left, top].indexOf(basePlacement) >= 0 ? -1 : 1;\n\n var _ref = typeof offset === 'function' ? offset(Object.assign({}, rects, {\n placement: placement\n })) : offset,\n skidding = _ref[0],\n distance = _ref[1];\n\n skidding = skidding || 0;\n distance = (distance || 0) * invertDistance;\n return [left, right].indexOf(basePlacement) >= 0 ? {\n x: distance,\n y: skidding\n } : {\n x: skidding,\n y: distance\n };\n}\n\nfunction offset(_ref2) {\n var state = _ref2.state,\n options = _ref2.options,\n name = _ref2.name;\n var _options$offset = options.offset,\n offset = _options$offset === void 0 ? [0, 0] : _options$offset;\n var data = placements.reduce(function (acc, placement) {\n acc[placement] = distanceAndSkiddingToXY(placement, state.rects, offset);\n return acc;\n }, {});\n var _data$state$placement = data[state.placement],\n x = _data$state$placement.x,\n y = _data$state$placement.y;\n\n if (state.modifiersData.popperOffsets != null) {\n state.modifiersData.popperOffsets.x += x;\n state.modifiersData.popperOffsets.y += y;\n }\n\n state.modifiersData[name] = data;\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'offset',\n enabled: true,\n phase: 'main',\n requires: ['popperOffsets'],\n fn: offset\n};","import computeOffsets from \"../utils/computeOffsets.js\";\n\nfunction popperOffsets(_ref) {\n var state = _ref.state,\n name = _ref.name;\n // Offsets are the actual position the popper needs to have to be\n // properly positioned near its reference element\n // This is the most basic placement, and will be adjusted by\n // the modifiers in the next step\n state.modifiersData[name] = computeOffsets({\n reference: state.rects.reference,\n element: state.rects.popper,\n strategy: 'absolute',\n placement: state.placement\n });\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'popperOffsets',\n enabled: true,\n phase: 'read',\n fn: popperOffsets,\n data: {}\n};","import { top, left, right, bottom, start } from \"../enums.js\";\nimport getBasePlacement from \"../utils/getBasePlacement.js\";\nimport getMainAxisFromPlacement from \"../utils/getMainAxisFromPlacement.js\";\nimport getAltAxis from \"../utils/getAltAxis.js\";\nimport { within, withinMaxClamp } from \"../utils/within.js\";\nimport getLayoutRect from \"../dom-utils/getLayoutRect.js\";\nimport getOffsetParent from \"../dom-utils/getOffsetParent.js\";\nimport detectOverflow from \"../utils/detectOverflow.js\";\nimport getVariation from \"../utils/getVariation.js\";\nimport getFreshSideObject from \"../utils/getFreshSideObject.js\";\nimport { min as mathMin, max as mathMax } from \"../utils/math.js\";\n\nfunction preventOverflow(_ref) {\n var state = _ref.state,\n options = _ref.options,\n name = _ref.name;\n var _options$mainAxis = options.mainAxis,\n checkMainAxis = _options$mainAxis === void 0 ? true : _options$mainAxis,\n _options$altAxis = options.altAxis,\n checkAltAxis = _options$altAxis === void 0 ? false : _options$altAxis,\n boundary = options.boundary,\n rootBoundary = options.rootBoundary,\n altBoundary = options.altBoundary,\n padding = options.padding,\n _options$tether = options.tether,\n tether = _options$tether === void 0 ? true : _options$tether,\n _options$tetherOffset = options.tetherOffset,\n tetherOffset = _options$tetherOffset === void 0 ? 0 : _options$tetherOffset;\n var overflow = detectOverflow(state, {\n boundary: boundary,\n rootBoundary: rootBoundary,\n padding: padding,\n altBoundary: altBoundary\n });\n var basePlacement = getBasePlacement(state.placement);\n var variation = getVariation(state.placement);\n var isBasePlacement = !variation;\n var mainAxis = getMainAxisFromPlacement(basePlacement);\n var altAxis = getAltAxis(mainAxis);\n var popperOffsets = state.modifiersData.popperOffsets;\n var referenceRect = state.rects.reference;\n var popperRect = state.rects.popper;\n var tetherOffsetValue = typeof tetherOffset === 'function' ? tetherOffset(Object.assign({}, state.rects, {\n placement: state.placement\n })) : tetherOffset;\n var normalizedTetherOffsetValue = typeof tetherOffsetValue === 'number' ? {\n mainAxis: tetherOffsetValue,\n altAxis: tetherOffsetValue\n } : Object.assign({\n mainAxis: 0,\n altAxis: 0\n }, tetherOffsetValue);\n var offsetModifierState = state.modifiersData.offset ? state.modifiersData.offset[state.placement] : null;\n var data = {\n x: 0,\n y: 0\n };\n\n if (!popperOffsets) {\n return;\n }\n\n if (checkMainAxis) {\n var _offsetModifierState$;\n\n var mainSide = mainAxis === 'y' ? top : left;\n var altSide = mainAxis === 'y' ? bottom : right;\n var len = mainAxis === 'y' ? 'height' : 'width';\n var offset = popperOffsets[mainAxis];\n var min = offset + overflow[mainSide];\n var max = offset - overflow[altSide];\n var additive = tether ? -popperRect[len] / 2 : 0;\n var minLen = variation === start ? referenceRect[len] : popperRect[len];\n var maxLen = variation === start ? -popperRect[len] : -referenceRect[len]; // We need to include the arrow in the calculation so the arrow doesn't go\n // outside the reference bounds\n\n var arrowElement = state.elements.arrow;\n var arrowRect = tether && arrowElement ? getLayoutRect(arrowElement) : {\n width: 0,\n height: 0\n };\n var arrowPaddingObject = state.modifiersData['arrow#persistent'] ? state.modifiersData['arrow#persistent'].padding : getFreshSideObject();\n var arrowPaddingMin = arrowPaddingObject[mainSide];\n var arrowPaddingMax = arrowPaddingObject[altSide]; // If the reference length is smaller than the arrow length, we don't want\n // to include its full size in the calculation. If the reference is small\n // and near the edge of a boundary, the popper can overflow even if the\n // reference is not overflowing as well (e.g. virtual elements with no\n // width or height)\n\n var arrowLen = within(0, referenceRect[len], arrowRect[len]);\n var minOffset = isBasePlacement ? referenceRect[len] / 2 - additive - arrowLen - arrowPaddingMin - normalizedTetherOffsetValue.mainAxis : minLen - arrowLen - arrowPaddingMin - normalizedTetherOffsetValue.mainAxis;\n var maxOffset = isBasePlacement ? -referenceRect[len] / 2 + additive + arrowLen + arrowPaddingMax + normalizedTetherOffsetValue.mainAxis : maxLen + arrowLen + arrowPaddingMax + normalizedTetherOffsetValue.mainAxis;\n var arrowOffsetParent = state.elements.arrow && getOffsetParent(state.elements.arrow);\n var clientOffset = arrowOffsetParent ? mainAxis === 'y' ? arrowOffsetParent.clientTop || 0 : arrowOffsetParent.clientLeft || 0 : 0;\n var offsetModifierValue = (_offsetModifierState$ = offsetModifierState == null ? void 0 : offsetModifierState[mainAxis]) != null ? _offsetModifierState$ : 0;\n var tetherMin = offset + minOffset - offsetModifierValue - clientOffset;\n var tetherMax = offset + maxOffset - offsetModifierValue;\n var preventedOffset = within(tether ? mathMin(min, tetherMin) : min, offset, tether ? mathMax(max, tetherMax) : max);\n popperOffsets[mainAxis] = preventedOffset;\n data[mainAxis] = preventedOffset - offset;\n }\n\n if (checkAltAxis) {\n var _offsetModifierState$2;\n\n var _mainSide = mainAxis === 'x' ? top : left;\n\n var _altSide = mainAxis === 'x' ? bottom : right;\n\n var _offset = popperOffsets[altAxis];\n\n var _len = altAxis === 'y' ? 'height' : 'width';\n\n var _min = _offset + overflow[_mainSide];\n\n var _max = _offset - overflow[_altSide];\n\n var isOriginSide = [top, left].indexOf(basePlacement) !== -1;\n\n var _offsetModifierValue = (_offsetModifierState$2 = offsetModifierState == null ? void 0 : offsetModifierState[altAxis]) != null ? _offsetModifierState$2 : 0;\n\n var _tetherMin = isOriginSide ? _min : _offset - referenceRect[_len] - popperRect[_len] - _offsetModifierValue + normalizedTetherOffsetValue.altAxis;\n\n var _tetherMax = isOriginSide ? _offset + referenceRect[_len] + popperRect[_len] - _offsetModifierValue - normalizedTetherOffsetValue.altAxis : _max;\n\n var _preventedOffset = tether && isOriginSide ? withinMaxClamp(_tetherMin, _offset, _tetherMax) : within(tether ? _tetherMin : _min, _offset, tether ? _tetherMax : _max);\n\n popperOffsets[altAxis] = _preventedOffset;\n data[altAxis] = _preventedOffset - _offset;\n }\n\n state.modifiersData[name] = data;\n} // eslint-disable-next-line import/no-unused-modules\n\n\nexport default {\n name: 'preventOverflow',\n enabled: true,\n phase: 'main',\n fn: preventOverflow,\n requiresIfExists: ['offset']\n};","export default function getAltAxis(axis) {\n return axis === 'x' ? 'y' : 'x';\n}","import getBoundingClientRect from \"./getBoundingClientRect.js\";\nimport getNodeScroll from \"./getNodeScroll.js\";\nimport getNodeName from \"./getNodeName.js\";\nimport { isHTMLElement } from \"./instanceOf.js\";\nimport getWindowScrollBarX from \"./getWindowScrollBarX.js\";\nimport getDocumentElement from \"./getDocumentElement.js\";\nimport isScrollParent from \"./isScrollParent.js\";\nimport { round } from \"../utils/math.js\";\n\nfunction isElementScaled(element) {\n var rect = element.getBoundingClientRect();\n var scaleX = round(rect.width) / element.offsetWidth || 1;\n var scaleY = round(rect.height) / element.offsetHeight || 1;\n return scaleX !== 1 || scaleY !== 1;\n} // Returns the composite rect of an element relative to its offsetParent.\n// Composite means it takes into account transforms as well as layout.\n\n\nexport default function getCompositeRect(elementOrVirtualElement, offsetParent, isFixed) {\n if (isFixed === void 0) {\n isFixed = false;\n }\n\n var isOffsetParentAnElement = isHTMLElement(offsetParent);\n var offsetParentIsScaled = isHTMLElement(offsetParent) && isElementScaled(offsetParent);\n var documentElement = getDocumentElement(offsetParent);\n var rect = getBoundingClientRect(elementOrVirtualElement, offsetParentIsScaled, isFixed);\n var scroll = {\n scrollLeft: 0,\n scrollTop: 0\n };\n var offsets = {\n x: 0,\n y: 0\n };\n\n if (isOffsetParentAnElement || !isOffsetParentAnElement && !isFixed) {\n if (getNodeName(offsetParent) !== 'body' || // https://github.com/popperjs/popper-core/issues/1078\n isScrollParent(documentElement)) {\n scroll = getNodeScroll(offsetParent);\n }\n\n if (isHTMLElement(offsetParent)) {\n offsets = getBoundingClientRect(offsetParent, true);\n offsets.x += offsetParent.clientLeft;\n offsets.y += offsetParent.clientTop;\n } else if (documentElement) {\n offsets.x = getWindowScrollBarX(documentElement);\n }\n }\n\n return {\n x: rect.left + scroll.scrollLeft - offsets.x,\n y: rect.top + scroll.scrollTop - offsets.y,\n width: rect.width,\n height: rect.height\n };\n}","import getWindowScroll from \"./getWindowScroll.js\";\nimport getWindow from \"./getWindow.js\";\nimport { isHTMLElement } from \"./instanceOf.js\";\nimport getHTMLElementScroll from \"./getHTMLElementScroll.js\";\nexport default function getNodeScroll(node) {\n if (node === getWindow(node) || !isHTMLElement(node)) {\n return getWindowScroll(node);\n } else {\n return getHTMLElementScroll(node);\n }\n}","export default function getHTMLElementScroll(element) {\n return {\n scrollLeft: element.scrollLeft,\n scrollTop: element.scrollTop\n };\n}","import { modifierPhases } from \"../enums.js\"; // source: https://stackoverflow.com/questions/49875255\n\nfunction order(modifiers) {\n var map = new Map();\n var visited = new Set();\n var result = [];\n modifiers.forEach(function (modifier) {\n map.set(modifier.name, modifier);\n }); // On visiting object, check for its dependencies and visit them recursively\n\n function sort(modifier) {\n visited.add(modifier.name);\n var requires = [].concat(modifier.requires || [], modifier.requiresIfExists || []);\n requires.forEach(function (dep) {\n if (!visited.has(dep)) {\n var depModifier = map.get(dep);\n\n if (depModifier) {\n sort(depModifier);\n }\n }\n });\n result.push(modifier);\n }\n\n modifiers.forEach(function (modifier) {\n if (!visited.has(modifier.name)) {\n // check for visited object\n sort(modifier);\n }\n });\n return result;\n}\n\nexport default function orderModifiers(modifiers) {\n // order based on dependencies\n var orderedModifiers = order(modifiers); // order based on phase\n\n return modifierPhases.reduce(function (acc, phase) {\n return acc.concat(orderedModifiers.filter(function (modifier) {\n return modifier.phase === phase;\n }));\n }, []);\n}","import getCompositeRect from \"./dom-utils/getCompositeRect.js\";\nimport getLayoutRect from \"./dom-utils/getLayoutRect.js\";\nimport listScrollParents from \"./dom-utils/listScrollParents.js\";\nimport getOffsetParent from \"./dom-utils/getOffsetParent.js\";\nimport orderModifiers from \"./utils/orderModifiers.js\";\nimport debounce from \"./utils/debounce.js\";\nimport mergeByName from \"./utils/mergeByName.js\";\nimport detectOverflow from \"./utils/detectOverflow.js\";\nimport { isElement } from \"./dom-utils/instanceOf.js\";\nvar DEFAULT_OPTIONS = {\n placement: 'bottom',\n modifiers: [],\n strategy: 'absolute'\n};\n\nfunction areValidElements() {\n for (var _len = arguments.length, args = new Array(_len), _key = 0; _key < _len; _key++) {\n args[_key] = arguments[_key];\n }\n\n return !args.some(function (element) {\n return !(element && typeof element.getBoundingClientRect === 'function');\n });\n}\n\nexport function popperGenerator(generatorOptions) {\n if (generatorOptions === void 0) {\n generatorOptions = {};\n }\n\n var _generatorOptions = generatorOptions,\n _generatorOptions$def = _generatorOptions.defaultModifiers,\n defaultModifiers = _generatorOptions$def === void 0 ? [] : _generatorOptions$def,\n _generatorOptions$def2 = _generatorOptions.defaultOptions,\n defaultOptions = _generatorOptions$def2 === void 0 ? DEFAULT_OPTIONS : _generatorOptions$def2;\n return function createPopper(reference, popper, options) {\n if (options === void 0) {\n options = defaultOptions;\n }\n\n var state = {\n placement: 'bottom',\n orderedModifiers: [],\n options: Object.assign({}, DEFAULT_OPTIONS, defaultOptions),\n modifiersData: {},\n elements: {\n reference: reference,\n popper: popper\n },\n attributes: {},\n styles: {}\n };\n var effectCleanupFns = [];\n var isDestroyed = false;\n var instance = {\n state: state,\n setOptions: function setOptions(setOptionsAction) {\n var options = typeof setOptionsAction === 'function' ? setOptionsAction(state.options) : setOptionsAction;\n cleanupModifierEffects();\n state.options = Object.assign({}, defaultOptions, state.options, options);\n state.scrollParents = {\n reference: isElement(reference) ? listScrollParents(reference) : reference.contextElement ? listScrollParents(reference.contextElement) : [],\n popper: listScrollParents(popper)\n }; // Orders the modifiers based on their dependencies and `phase`\n // properties\n\n var orderedModifiers = orderModifiers(mergeByName([].concat(defaultModifiers, state.options.modifiers))); // Strip out disabled modifiers\n\n state.orderedModifiers = orderedModifiers.filter(function (m) {\n return m.enabled;\n });\n runModifierEffects();\n return instance.update();\n },\n // Sync update – it will always be executed, even if not necessary. This\n // is useful for low frequency updates where sync behavior simplifies the\n // logic.\n // For high frequency updates (e.g. `resize` and `scroll` events), always\n // prefer the async Popper#update method\n forceUpdate: function forceUpdate() {\n if (isDestroyed) {\n return;\n }\n\n var _state$elements = state.elements,\n reference = _state$elements.reference,\n popper = _state$elements.popper; // Don't proceed if `reference` or `popper` are not valid elements\n // anymore\n\n if (!areValidElements(reference, popper)) {\n return;\n } // Store the reference and popper rects to be read by modifiers\n\n\n state.rects = {\n reference: getCompositeRect(reference, getOffsetParent(popper), state.options.strategy === 'fixed'),\n popper: getLayoutRect(popper)\n }; // Modifiers have the ability to reset the current update cycle. The\n // most common use case for this is the `flip` modifier changing the\n // placement, which then needs to re-run all the modifiers, because the\n // logic was previously ran for the previous placement and is therefore\n // stale/incorrect\n\n state.reset = false;\n state.placement = state.options.placement; // On each update cycle, the `modifiersData` property for each modifier\n // is filled with the initial data specified by the modifier. This means\n // it doesn't persist and is fresh on each update.\n // To ensure persistent data, use `${name}#persistent`\n\n state.orderedModifiers.forEach(function (modifier) {\n return state.modifiersData[modifier.name] = Object.assign({}, modifier.data);\n });\n\n for (var index = 0; index < state.orderedModifiers.length; index++) {\n if (state.reset === true) {\n state.reset = false;\n index = -1;\n continue;\n }\n\n var _state$orderedModifie = state.orderedModifiers[index],\n fn = _state$orderedModifie.fn,\n _state$orderedModifie2 = _state$orderedModifie.options,\n _options = _state$orderedModifie2 === void 0 ? {} : _state$orderedModifie2,\n name = _state$orderedModifie.name;\n\n if (typeof fn === 'function') {\n state = fn({\n state: state,\n options: _options,\n name: name,\n instance: instance\n }) || state;\n }\n }\n },\n // Async and optimistically optimized update – it will not be executed if\n // not necessary (debounced to run at most once-per-tick)\n update: debounce(function () {\n return new Promise(function (resolve) {\n instance.forceUpdate();\n resolve(state);\n });\n }),\n destroy: function destroy() {\n cleanupModifierEffects();\n isDestroyed = true;\n }\n };\n\n if (!areValidElements(reference, popper)) {\n return instance;\n }\n\n instance.setOptions(options).then(function (state) {\n if (!isDestroyed && options.onFirstUpdate) {\n options.onFirstUpdate(state);\n }\n }); // Modifiers have the ability to execute arbitrary code before the first\n // update cycle runs. They will be executed in the same order as the update\n // cycle. This is useful when a modifier adds some persistent data that\n // other modifiers need to use, but the modifier is run after the dependent\n // one.\n\n function runModifierEffects() {\n state.orderedModifiers.forEach(function (_ref) {\n var name = _ref.name,\n _ref$options = _ref.options,\n options = _ref$options === void 0 ? {} : _ref$options,\n effect = _ref.effect;\n\n if (typeof effect === 'function') {\n var cleanupFn = effect({\n state: state,\n name: name,\n instance: instance,\n options: options\n });\n\n var noopFn = function noopFn() {};\n\n effectCleanupFns.push(cleanupFn || noopFn);\n }\n });\n }\n\n function cleanupModifierEffects() {\n effectCleanupFns.forEach(function (fn) {\n return fn();\n });\n effectCleanupFns = [];\n }\n\n return instance;\n };\n}\nexport var createPopper = /*#__PURE__*/popperGenerator(); // eslint-disable-next-line import/no-unused-modules\n\nexport { detectOverflow };","export default function debounce(fn) {\n var pending;\n return function () {\n if (!pending) {\n pending = new Promise(function (resolve) {\n Promise.resolve().then(function () {\n pending = undefined;\n resolve(fn());\n });\n });\n }\n\n return pending;\n };\n}","export default function mergeByName(modifiers) {\n var merged = modifiers.reduce(function (merged, current) {\n var existing = merged[current.name];\n merged[current.name] = existing ? Object.assign({}, existing, current, {\n options: Object.assign({}, existing.options, current.options),\n data: Object.assign({}, existing.data, current.data)\n }) : current;\n return merged;\n }, {}); // IE11 does not support Object.values\n\n return Object.keys(merged).map(function (key) {\n return merged[key];\n });\n}","import { popperGenerator, detectOverflow } from \"./createPopper.js\";\nimport eventListeners from \"./modifiers/eventListeners.js\";\nimport popperOffsets from \"./modifiers/popperOffsets.js\";\nimport computeStyles from \"./modifiers/computeStyles.js\";\nimport applyStyles from \"./modifiers/applyStyles.js\";\nvar defaultModifiers = [eventListeners, popperOffsets, computeStyles, applyStyles];\nvar createPopper = /*#__PURE__*/popperGenerator({\n defaultModifiers: defaultModifiers\n}); // eslint-disable-next-line import/no-unused-modules\n\nexport { createPopper, popperGenerator, defaultModifiers, detectOverflow };","import { popperGenerator, detectOverflow } from \"./createPopper.js\";\nimport eventListeners from \"./modifiers/eventListeners.js\";\nimport popperOffsets from \"./modifiers/popperOffsets.js\";\nimport computeStyles from \"./modifiers/computeStyles.js\";\nimport applyStyles from \"./modifiers/applyStyles.js\";\nimport offset from \"./modifiers/offset.js\";\nimport flip from \"./modifiers/flip.js\";\nimport preventOverflow from \"./modifiers/preventOverflow.js\";\nimport arrow from \"./modifiers/arrow.js\";\nimport hide from \"./modifiers/hide.js\";\nvar defaultModifiers = [eventListeners, popperOffsets, computeStyles, applyStyles, offset, flip, preventOverflow, arrow, hide];\nvar createPopper = /*#__PURE__*/popperGenerator({\n defaultModifiers: defaultModifiers\n}); // eslint-disable-next-line import/no-unused-modules\n\nexport { createPopper, popperGenerator, defaultModifiers, detectOverflow }; // eslint-disable-next-line import/no-unused-modules\n\nexport { createPopper as createPopperLite } from \"./popper-lite.js\"; // eslint-disable-next-line import/no-unused-modules\n\nexport * from \"./modifiers/index.js\";","/**\n * --------------------------------------------------------------------------\n * Bootstrap dropdown.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport * as Popper from '@popperjs/core'\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport Manipulator from './dom/manipulator.js'\nimport SelectorEngine from './dom/selector-engine.js'\nimport {\n defineJQueryPlugin,\n execute,\n getElement,\n getNextActiveElement,\n isDisabled,\n isElement,\n isRTL,\n isVisible,\n noop\n} from './util/index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'dropdown'\nconst DATA_KEY = 'bs.dropdown'\nconst EVENT_KEY = `.${DATA_KEY}`\nconst DATA_API_KEY = '.data-api'\n\nconst ESCAPE_KEY = 'Escape'\nconst TAB_KEY = 'Tab'\nconst ARROW_UP_KEY = 'ArrowUp'\nconst ARROW_DOWN_KEY = 'ArrowDown'\nconst RIGHT_MOUSE_BUTTON = 2 // MouseEvent.button value for the secondary button, usually the right button\n\nconst EVENT_HIDE = `hide${EVENT_KEY}`\nconst EVENT_HIDDEN = `hidden${EVENT_KEY}`\nconst EVENT_SHOW = `show${EVENT_KEY}`\nconst EVENT_SHOWN = `shown${EVENT_KEY}`\nconst EVENT_CLICK_DATA_API = `click${EVENT_KEY}${DATA_API_KEY}`\nconst EVENT_KEYDOWN_DATA_API = `keydown${EVENT_KEY}${DATA_API_KEY}`\nconst EVENT_KEYUP_DATA_API = `keyup${EVENT_KEY}${DATA_API_KEY}`\n\nconst CLASS_NAME_SHOW = 'show'\nconst CLASS_NAME_DROPUP = 'dropup'\nconst CLASS_NAME_DROPEND = 'dropend'\nconst CLASS_NAME_DROPSTART = 'dropstart'\nconst CLASS_NAME_DROPUP_CENTER = 'dropup-center'\nconst CLASS_NAME_DROPDOWN_CENTER = 'dropdown-center'\n\nconst SELECTOR_DATA_TOGGLE = '[data-bs-toggle=\"dropdown\"]:not(.disabled):not(:disabled)'\nconst SELECTOR_DATA_TOGGLE_SHOWN = `${SELECTOR_DATA_TOGGLE}.${CLASS_NAME_SHOW}`\nconst SELECTOR_MENU = '.dropdown-menu'\nconst SELECTOR_NAVBAR = '.navbar'\nconst SELECTOR_NAVBAR_NAV = '.navbar-nav'\nconst SELECTOR_VISIBLE_ITEMS = '.dropdown-menu .dropdown-item:not(.disabled):not(:disabled)'\n\nconst PLACEMENT_TOP = isRTL() ? 'top-end' : 'top-start'\nconst PLACEMENT_TOPEND = isRTL() ? 'top-start' : 'top-end'\nconst PLACEMENT_BOTTOM = isRTL() ? 'bottom-end' : 'bottom-start'\nconst PLACEMENT_BOTTOMEND = isRTL() ? 'bottom-start' : 'bottom-end'\nconst PLACEMENT_RIGHT = isRTL() ? 'left-start' : 'right-start'\nconst PLACEMENT_LEFT = isRTL() ? 'right-start' : 'left-start'\nconst PLACEMENT_TOPCENTER = 'top'\nconst PLACEMENT_BOTTOMCENTER = 'bottom'\n\nconst Default = {\n autoClose: true,\n boundary: 'clippingParents',\n display: 'dynamic',\n offset: [0, 2],\n popperConfig: null,\n reference: 'toggle'\n}\n\nconst DefaultType = {\n autoClose: '(boolean|string)',\n boundary: '(string|element)',\n display: 'string',\n offset: '(array|string|function)',\n popperConfig: '(null|object|function)',\n reference: '(string|element|object)'\n}\n\n/**\n * Class definition\n */\n\nclass Dropdown extends BaseComponent {\n constructor(element, config) {\n super(element, config)\n\n this._popper = null\n this._parent = this._element.parentNode // dropdown wrapper\n // TODO: v6 revert #37011 & change markup https://getbootstrap.com/docs/5.3/forms/input-group/\n this._menu = SelectorEngine.next(this._element, SELECTOR_MENU)[0] ||\n SelectorEngine.prev(this._element, SELECTOR_MENU)[0] ||\n SelectorEngine.findOne(SELECTOR_MENU, this._parent)\n this._inNavbar = this._detectNavbar()\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n toggle() {\n return this._isShown() ? this.hide() : this.show()\n }\n\n show() {\n if (isDisabled(this._element) || this._isShown()) {\n return\n }\n\n const relatedTarget = {\n relatedTarget: this._element\n }\n\n const showEvent = EventHandler.trigger(this._element, EVENT_SHOW, relatedTarget)\n\n if (showEvent.defaultPrevented) {\n return\n }\n\n this._createPopper()\n\n // If this is a touch-enabled device we add extra\n // empty mouseover listeners to the body's immediate children;\n // only needed because of broken event delegation on iOS\n // https://www.quirksmode.org/blog/archives/2014/02/mouse_event_bub.html\n if ('ontouchstart' in document.documentElement && !this._parent.closest(SELECTOR_NAVBAR_NAV)) {\n for (const element of [].concat(...document.body.children)) {\n EventHandler.on(element, 'mouseover', noop)\n }\n }\n\n this._element.focus()\n this._element.setAttribute('aria-expanded', true)\n\n this._menu.classList.add(CLASS_NAME_SHOW)\n this._element.classList.add(CLASS_NAME_SHOW)\n EventHandler.trigger(this._element, EVENT_SHOWN, relatedTarget)\n }\n\n hide() {\n if (isDisabled(this._element) || !this._isShown()) {\n return\n }\n\n const relatedTarget = {\n relatedTarget: this._element\n }\n\n this._completeHide(relatedTarget)\n }\n\n dispose() {\n if (this._popper) {\n this._popper.destroy()\n }\n\n super.dispose()\n }\n\n update() {\n this._inNavbar = this._detectNavbar()\n if (this._popper) {\n this._popper.update()\n }\n }\n\n // Private\n _completeHide(relatedTarget) {\n const hideEvent = EventHandler.trigger(this._element, EVENT_HIDE, relatedTarget)\n if (hideEvent.defaultPrevented) {\n return\n }\n\n // If this is a touch-enabled device we remove the extra\n // empty mouseover listeners we added for iOS support\n if ('ontouchstart' in document.documentElement) {\n for (const element of [].concat(...document.body.children)) {\n EventHandler.off(element, 'mouseover', noop)\n }\n }\n\n if (this._popper) {\n this._popper.destroy()\n }\n\n this._menu.classList.remove(CLASS_NAME_SHOW)\n this._element.classList.remove(CLASS_NAME_SHOW)\n this._element.setAttribute('aria-expanded', 'false')\n Manipulator.removeDataAttribute(this._menu, 'popper')\n EventHandler.trigger(this._element, EVENT_HIDDEN, relatedTarget)\n }\n\n _getConfig(config) {\n config = super._getConfig(config)\n\n if (typeof config.reference === 'object' && !isElement(config.reference) &&\n typeof config.reference.getBoundingClientRect !== 'function'\n ) {\n // Popper virtual elements require a getBoundingClientRect method\n throw new TypeError(`${NAME.toUpperCase()}: Option \"reference\" provided type \"object\" without a required \"getBoundingClientRect\" method.`)\n }\n\n return config\n }\n\n _createPopper() {\n if (typeof Popper === 'undefined') {\n throw new TypeError('Bootstrap\\'s dropdowns require Popper (https://popper.js.org)')\n }\n\n let referenceElement = this._element\n\n if (this._config.reference === 'parent') {\n referenceElement = this._parent\n } else if (isElement(this._config.reference)) {\n referenceElement = getElement(this._config.reference)\n } else if (typeof this._config.reference === 'object') {\n referenceElement = this._config.reference\n }\n\n const popperConfig = this._getPopperConfig()\n this._popper = Popper.createPopper(referenceElement, this._menu, popperConfig)\n }\n\n _isShown() {\n return this._menu.classList.contains(CLASS_NAME_SHOW)\n }\n\n _getPlacement() {\n const parentDropdown = this._parent\n\n if (parentDropdown.classList.contains(CLASS_NAME_DROPEND)) {\n return PLACEMENT_RIGHT\n }\n\n if (parentDropdown.classList.contains(CLASS_NAME_DROPSTART)) {\n return PLACEMENT_LEFT\n }\n\n if (parentDropdown.classList.contains(CLASS_NAME_DROPUP_CENTER)) {\n return PLACEMENT_TOPCENTER\n }\n\n if (parentDropdown.classList.contains(CLASS_NAME_DROPDOWN_CENTER)) {\n return PLACEMENT_BOTTOMCENTER\n }\n\n // We need to trim the value because custom properties can also include spaces\n const isEnd = getComputedStyle(this._menu).getPropertyValue('--bs-position').trim() === 'end'\n\n if (parentDropdown.classList.contains(CLASS_NAME_DROPUP)) {\n return isEnd ? PLACEMENT_TOPEND : PLACEMENT_TOP\n }\n\n return isEnd ? PLACEMENT_BOTTOMEND : PLACEMENT_BOTTOM\n }\n\n _detectNavbar() {\n return this._element.closest(SELECTOR_NAVBAR) !== null\n }\n\n _getOffset() {\n const { offset } = this._config\n\n if (typeof offset === 'string') {\n return offset.split(',').map(value => Number.parseInt(value, 10))\n }\n\n if (typeof offset === 'function') {\n return popperData => offset(popperData, this._element)\n }\n\n return offset\n }\n\n _getPopperConfig() {\n const defaultBsPopperConfig = {\n placement: this._getPlacement(),\n modifiers: [{\n name: 'preventOverflow',\n options: {\n boundary: this._config.boundary\n }\n },\n {\n name: 'offset',\n options: {\n offset: this._getOffset()\n }\n }]\n }\n\n // Disable Popper if we have a static display or Dropdown is in Navbar\n if (this._inNavbar || this._config.display === 'static') {\n Manipulator.setDataAttribute(this._menu, 'popper', 'static') // TODO: v6 remove\n defaultBsPopperConfig.modifiers = [{\n name: 'applyStyles',\n enabled: false\n }]\n }\n\n return {\n ...defaultBsPopperConfig,\n ...execute(this._config.popperConfig, [defaultBsPopperConfig])\n }\n }\n\n _selectMenuItem({ key, target }) {\n const items = SelectorEngine.find(SELECTOR_VISIBLE_ITEMS, this._menu).filter(element => isVisible(element))\n\n if (!items.length) {\n return\n }\n\n // if target isn't included in items (e.g. when expanding the dropdown)\n // allow cycling to get the last item in case key equals ARROW_UP_KEY\n getNextActiveElement(items, target, key === ARROW_DOWN_KEY, !items.includes(target)).focus()\n }\n\n // Static\n static jQueryInterface(config) {\n return this.each(function () {\n const data = Dropdown.getOrCreateInstance(this, config)\n\n if (typeof config !== 'string') {\n return\n }\n\n if (typeof data[config] === 'undefined') {\n throw new TypeError(`No method named \"${config}\"`)\n }\n\n data[config]()\n })\n }\n\n static clearMenus(event) {\n if (event.button === RIGHT_MOUSE_BUTTON || (event.type === 'keyup' && event.key !== TAB_KEY)) {\n return\n }\n\n const openToggles = SelectorEngine.find(SELECTOR_DATA_TOGGLE_SHOWN)\n\n for (const toggle of openToggles) {\n const context = Dropdown.getInstance(toggle)\n if (!context || context._config.autoClose === false) {\n continue\n }\n\n const composedPath = event.composedPath()\n const isMenuTarget = composedPath.includes(context._menu)\n if (\n composedPath.includes(context._element) ||\n (context._config.autoClose === 'inside' && !isMenuTarget) ||\n (context._config.autoClose === 'outside' && isMenuTarget)\n ) {\n continue\n }\n\n // Tab navigation through the dropdown menu or events from contained inputs shouldn't close the menu\n if (context._menu.contains(event.target) && ((event.type === 'keyup' && event.key === TAB_KEY) || /input|select|option|textarea|form/i.test(event.target.tagName))) {\n continue\n }\n\n const relatedTarget = { relatedTarget: context._element }\n\n if (event.type === 'click') {\n relatedTarget.clickEvent = event\n }\n\n context._completeHide(relatedTarget)\n }\n }\n\n static dataApiKeydownHandler(event) {\n // If not an UP | DOWN | ESCAPE key => not a dropdown command\n // If input/textarea && if key is other than ESCAPE => not a dropdown command\n\n const isInput = /input|textarea/i.test(event.target.tagName)\n const isEscapeEvent = event.key === ESCAPE_KEY\n const isUpOrDownEvent = [ARROW_UP_KEY, ARROW_DOWN_KEY].includes(event.key)\n\n if (!isUpOrDownEvent && !isEscapeEvent) {\n return\n }\n\n if (isInput && !isEscapeEvent) {\n return\n }\n\n event.preventDefault()\n\n // TODO: v6 revert #37011 & change markup https://getbootstrap.com/docs/5.3/forms/input-group/\n const getToggleButton = this.matches(SELECTOR_DATA_TOGGLE) ?\n this :\n (SelectorEngine.prev(this, SELECTOR_DATA_TOGGLE)[0] ||\n SelectorEngine.next(this, SELECTOR_DATA_TOGGLE)[0] ||\n SelectorEngine.findOne(SELECTOR_DATA_TOGGLE, event.delegateTarget.parentNode))\n\n const instance = Dropdown.getOrCreateInstance(getToggleButton)\n\n if (isUpOrDownEvent) {\n event.stopPropagation()\n instance.show()\n instance._selectMenuItem(event)\n return\n }\n\n if (instance._isShown()) { // else is escape and we check if it is shown\n event.stopPropagation()\n instance.hide()\n getToggleButton.focus()\n }\n }\n}\n\n/**\n * Data API implementation\n */\n\nEventHandler.on(document, EVENT_KEYDOWN_DATA_API, SELECTOR_DATA_TOGGLE, Dropdown.dataApiKeydownHandler)\nEventHandler.on(document, EVENT_KEYDOWN_DATA_API, SELECTOR_MENU, Dropdown.dataApiKeydownHandler)\nEventHandler.on(document, EVENT_CLICK_DATA_API, Dropdown.clearMenus)\nEventHandler.on(document, EVENT_KEYUP_DATA_API, Dropdown.clearMenus)\nEventHandler.on(document, EVENT_CLICK_DATA_API, SELECTOR_DATA_TOGGLE, function (event) {\n event.preventDefault()\n Dropdown.getOrCreateInstance(this).toggle()\n})\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Dropdown)\n\nexport default Dropdown\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/backdrop.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport EventHandler from '../dom/event-handler.js'\nimport Config from './config.js'\nimport { execute, executeAfterTransition, getElement, reflow } from './index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'backdrop'\nconst CLASS_NAME_FADE = 'fade'\nconst CLASS_NAME_SHOW = 'show'\nconst EVENT_MOUSEDOWN = `mousedown.bs.${NAME}`\n\nconst Default = {\n className: 'modal-backdrop',\n clickCallback: null,\n isAnimated: false,\n isVisible: true, // if false, we use the backdrop helper without adding any element to the dom\n rootElement: 'body' // give the choice to place backdrop under different elements\n}\n\nconst DefaultType = {\n className: 'string',\n clickCallback: '(function|null)',\n isAnimated: 'boolean',\n isVisible: 'boolean',\n rootElement: '(element|string)'\n}\n\n/**\n * Class definition\n */\n\nclass Backdrop extends Config {\n constructor(config) {\n super()\n this._config = this._getConfig(config)\n this._isAppended = false\n this._element = null\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n show(callback) {\n if (!this._config.isVisible) {\n execute(callback)\n return\n }\n\n this._append()\n\n const element = this._getElement()\n if (this._config.isAnimated) {\n reflow(element)\n }\n\n element.classList.add(CLASS_NAME_SHOW)\n\n this._emulateAnimation(() => {\n execute(callback)\n })\n }\n\n hide(callback) {\n if (!this._config.isVisible) {\n execute(callback)\n return\n }\n\n this._getElement().classList.remove(CLASS_NAME_SHOW)\n\n this._emulateAnimation(() => {\n this.dispose()\n execute(callback)\n })\n }\n\n dispose() {\n if (!this._isAppended) {\n return\n }\n\n EventHandler.off(this._element, EVENT_MOUSEDOWN)\n\n this._element.remove()\n this._isAppended = false\n }\n\n // Private\n _getElement() {\n if (!this._element) {\n const backdrop = document.createElement('div')\n backdrop.className = this._config.className\n if (this._config.isAnimated) {\n backdrop.classList.add(CLASS_NAME_FADE)\n }\n\n this._element = backdrop\n }\n\n return this._element\n }\n\n _configAfterMerge(config) {\n // use getElement() with the default \"body\" to get a fresh Element on each instantiation\n config.rootElement = getElement(config.rootElement)\n return config\n }\n\n _append() {\n if (this._isAppended) {\n return\n }\n\n const element = this._getElement()\n this._config.rootElement.append(element)\n\n EventHandler.on(element, EVENT_MOUSEDOWN, () => {\n execute(this._config.clickCallback)\n })\n\n this._isAppended = true\n }\n\n _emulateAnimation(callback) {\n executeAfterTransition(callback, this._getElement(), this._config.isAnimated)\n }\n}\n\nexport default Backdrop\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/focustrap.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport EventHandler from '../dom/event-handler.js'\nimport SelectorEngine from '../dom/selector-engine.js'\nimport Config from './config.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'focustrap'\nconst DATA_KEY = 'bs.focustrap'\nconst EVENT_KEY = `.${DATA_KEY}`\nconst EVENT_FOCUSIN = `focusin${EVENT_KEY}`\nconst EVENT_KEYDOWN_TAB = `keydown.tab${EVENT_KEY}`\n\nconst TAB_KEY = 'Tab'\nconst TAB_NAV_FORWARD = 'forward'\nconst TAB_NAV_BACKWARD = 'backward'\n\nconst Default = {\n autofocus: true,\n trapElement: null // The element to trap focus inside of\n}\n\nconst DefaultType = {\n autofocus: 'boolean',\n trapElement: 'element'\n}\n\n/**\n * Class definition\n */\n\nclass FocusTrap extends Config {\n constructor(config) {\n super()\n this._config = this._getConfig(config)\n this._isActive = false\n this._lastTabNavDirection = null\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n activate() {\n if (this._isActive) {\n return\n }\n\n if (this._config.autofocus) {\n this._config.trapElement.focus()\n }\n\n EventHandler.off(document, EVENT_KEY) // guard against infinite focus loop\n EventHandler.on(document, EVENT_FOCUSIN, event => this._handleFocusin(event))\n EventHandler.on(document, EVENT_KEYDOWN_TAB, event => this._handleKeydown(event))\n\n this._isActive = true\n }\n\n deactivate() {\n if (!this._isActive) {\n return\n }\n\n this._isActive = false\n EventHandler.off(document, EVENT_KEY)\n }\n\n // Private\n _handleFocusin(event) {\n const { trapElement } = this._config\n\n if (event.target === document || event.target === trapElement || trapElement.contains(event.target)) {\n return\n }\n\n const elements = SelectorEngine.focusableChildren(trapElement)\n\n if (elements.length === 0) {\n trapElement.focus()\n } else if (this._lastTabNavDirection === TAB_NAV_BACKWARD) {\n elements[elements.length - 1].focus()\n } else {\n elements[0].focus()\n }\n }\n\n _handleKeydown(event) {\n if (event.key !== TAB_KEY) {\n return\n }\n\n this._lastTabNavDirection = event.shiftKey ? TAB_NAV_BACKWARD : TAB_NAV_FORWARD\n }\n}\n\nexport default FocusTrap\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/scrollBar.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport Manipulator from '../dom/manipulator.js'\nimport SelectorEngine from '../dom/selector-engine.js'\nimport { isElement } from './index.js'\n\n/**\n * Constants\n */\n\nconst SELECTOR_FIXED_CONTENT = '.fixed-top, .fixed-bottom, .is-fixed, .sticky-top'\nconst SELECTOR_STICKY_CONTENT = '.sticky-top'\nconst PROPERTY_PADDING = 'padding-right'\nconst PROPERTY_MARGIN = 'margin-right'\n\n/**\n * Class definition\n */\n\nclass ScrollBarHelper {\n constructor() {\n this._element = document.body\n }\n\n // Public\n getWidth() {\n // https://developer.mozilla.org/en-US/docs/Web/API/Window/innerWidth#usage_notes\n const documentWidth = document.documentElement.clientWidth\n return Math.abs(window.innerWidth - documentWidth)\n }\n\n hide() {\n const width = this.getWidth()\n this._disableOverFlow()\n // give padding to element to balance the hidden scrollbar width\n this._setElementAttributes(this._element, PROPERTY_PADDING, calculatedValue => calculatedValue + width)\n // trick: We adjust positive paddingRight and negative marginRight to sticky-top elements to keep showing fullwidth\n this._setElementAttributes(SELECTOR_FIXED_CONTENT, PROPERTY_PADDING, calculatedValue => calculatedValue + width)\n this._setElementAttributes(SELECTOR_STICKY_CONTENT, PROPERTY_MARGIN, calculatedValue => calculatedValue - width)\n }\n\n reset() {\n this._resetElementAttributes(this._element, 'overflow')\n this._resetElementAttributes(this._element, PROPERTY_PADDING)\n this._resetElementAttributes(SELECTOR_FIXED_CONTENT, PROPERTY_PADDING)\n this._resetElementAttributes(SELECTOR_STICKY_CONTENT, PROPERTY_MARGIN)\n }\n\n isOverflowing() {\n return this.getWidth() > 0\n }\n\n // Private\n _disableOverFlow() {\n this._saveInitialAttribute(this._element, 'overflow')\n this._element.style.overflow = 'hidden'\n }\n\n _setElementAttributes(selector, styleProperty, callback) {\n const scrollbarWidth = this.getWidth()\n const manipulationCallBack = element => {\n if (element !== this._element && window.innerWidth > element.clientWidth + scrollbarWidth) {\n return\n }\n\n this._saveInitialAttribute(element, styleProperty)\n const calculatedValue = window.getComputedStyle(element).getPropertyValue(styleProperty)\n element.style.setProperty(styleProperty, `${callback(Number.parseFloat(calculatedValue))}px`)\n }\n\n this._applyManipulationCallback(selector, manipulationCallBack)\n }\n\n _saveInitialAttribute(element, styleProperty) {\n const actualValue = element.style.getPropertyValue(styleProperty)\n if (actualValue) {\n Manipulator.setDataAttribute(element, styleProperty, actualValue)\n }\n }\n\n _resetElementAttributes(selector, styleProperty) {\n const manipulationCallBack = element => {\n const value = Manipulator.getDataAttribute(element, styleProperty)\n // We only want to remove the property if the value is `null`; the value can also be zero\n if (value === null) {\n element.style.removeProperty(styleProperty)\n return\n }\n\n Manipulator.removeDataAttribute(element, styleProperty)\n element.style.setProperty(styleProperty, value)\n }\n\n this._applyManipulationCallback(selector, manipulationCallBack)\n }\n\n _applyManipulationCallback(selector, callBack) {\n if (isElement(selector)) {\n callBack(selector)\n return\n }\n\n for (const sel of SelectorEngine.find(selector, this._element)) {\n callBack(sel)\n }\n }\n}\n\nexport default ScrollBarHelper\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap modal.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport SelectorEngine from './dom/selector-engine.js'\nimport Backdrop from './util/backdrop.js'\nimport { enableDismissTrigger } from './util/component-functions.js'\nimport FocusTrap from './util/focustrap.js'\nimport { defineJQueryPlugin, isRTL, isVisible, reflow } from './util/index.js'\nimport ScrollBarHelper from './util/scrollbar.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'modal'\nconst DATA_KEY = 'bs.modal'\nconst EVENT_KEY = `.${DATA_KEY}`\nconst DATA_API_KEY = '.data-api'\nconst ESCAPE_KEY = 'Escape'\n\nconst EVENT_HIDE = `hide${EVENT_KEY}`\nconst EVENT_HIDE_PREVENTED = `hidePrevented${EVENT_KEY}`\nconst EVENT_HIDDEN = `hidden${EVENT_KEY}`\nconst EVENT_SHOW = `show${EVENT_KEY}`\nconst EVENT_SHOWN = `shown${EVENT_KEY}`\nconst EVENT_RESIZE = `resize${EVENT_KEY}`\nconst EVENT_CLICK_DISMISS = `click.dismiss${EVENT_KEY}`\nconst EVENT_MOUSEDOWN_DISMISS = `mousedown.dismiss${EVENT_KEY}`\nconst EVENT_KEYDOWN_DISMISS = `keydown.dismiss${EVENT_KEY}`\nconst EVENT_CLICK_DATA_API = `click${EVENT_KEY}${DATA_API_KEY}`\n\nconst CLASS_NAME_OPEN = 'modal-open'\nconst CLASS_NAME_FADE = 'fade'\nconst CLASS_NAME_SHOW = 'show'\nconst CLASS_NAME_STATIC = 'modal-static'\n\nconst OPEN_SELECTOR = '.modal.show'\nconst SELECTOR_DIALOG = '.modal-dialog'\nconst SELECTOR_MODAL_BODY = '.modal-body'\nconst SELECTOR_DATA_TOGGLE = '[data-bs-toggle=\"modal\"]'\n\nconst Default = {\n backdrop: true,\n focus: true,\n keyboard: true\n}\n\nconst DefaultType = {\n backdrop: '(boolean|string)',\n focus: 'boolean',\n keyboard: 'boolean'\n}\n\n/**\n * Class definition\n */\n\nclass Modal extends BaseComponent {\n constructor(element, config) {\n super(element, config)\n\n this._dialog = SelectorEngine.findOne(SELECTOR_DIALOG, this._element)\n this._backdrop = this._initializeBackDrop()\n this._focustrap = this._initializeFocusTrap()\n this._isShown = false\n this._isTransitioning = false\n this._scrollBar = new ScrollBarHelper()\n\n this._addEventListeners()\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n toggle(relatedTarget) {\n return this._isShown ? this.hide() : this.show(relatedTarget)\n }\n\n show(relatedTarget) {\n if (this._isShown || this._isTransitioning) {\n return\n }\n\n const showEvent = EventHandler.trigger(this._element, EVENT_SHOW, {\n relatedTarget\n })\n\n if (showEvent.defaultPrevented) {\n return\n }\n\n this._isShown = true\n this._isTransitioning = true\n\n this._scrollBar.hide()\n\n document.body.classList.add(CLASS_NAME_OPEN)\n\n this._adjustDialog()\n\n this._backdrop.show(() => this._showElement(relatedTarget))\n }\n\n hide() {\n if (!this._isShown || this._isTransitioning) {\n return\n }\n\n const hideEvent = EventHandler.trigger(this._element, EVENT_HIDE)\n\n if (hideEvent.defaultPrevented) {\n return\n }\n\n this._isShown = false\n this._isTransitioning = true\n this._focustrap.deactivate()\n\n this._element.classList.remove(CLASS_NAME_SHOW)\n\n this._queueCallback(() => this._hideModal(), this._element, this._isAnimated())\n }\n\n dispose() {\n EventHandler.off(window, EVENT_KEY)\n EventHandler.off(this._dialog, EVENT_KEY)\n\n this._backdrop.dispose()\n this._focustrap.deactivate()\n\n super.dispose()\n }\n\n handleUpdate() {\n this._adjustDialog()\n }\n\n // Private\n _initializeBackDrop() {\n return new Backdrop({\n isVisible: Boolean(this._config.backdrop), // 'static' option will be translated to true, and booleans will keep their value,\n isAnimated: this._isAnimated()\n })\n }\n\n _initializeFocusTrap() {\n return new FocusTrap({\n trapElement: this._element\n })\n }\n\n _showElement(relatedTarget) {\n // try to append dynamic modal\n if (!document.body.contains(this._element)) {\n document.body.append(this._element)\n }\n\n this._element.style.display = 'block'\n this._element.removeAttribute('aria-hidden')\n this._element.setAttribute('aria-modal', true)\n this._element.setAttribute('role', 'dialog')\n this._element.scrollTop = 0\n\n const modalBody = SelectorEngine.findOne(SELECTOR_MODAL_BODY, this._dialog)\n if (modalBody) {\n modalBody.scrollTop = 0\n }\n\n reflow(this._element)\n\n this._element.classList.add(CLASS_NAME_SHOW)\n\n const transitionComplete = () => {\n if (this._config.focus) {\n this._focustrap.activate()\n }\n\n this._isTransitioning = false\n EventHandler.trigger(this._element, EVENT_SHOWN, {\n relatedTarget\n })\n }\n\n this._queueCallback(transitionComplete, this._dialog, this._isAnimated())\n }\n\n _addEventListeners() {\n EventHandler.on(this._element, EVENT_KEYDOWN_DISMISS, event => {\n if (event.key !== ESCAPE_KEY) {\n return\n }\n\n if (this._config.keyboard) {\n this.hide()\n return\n }\n\n this._triggerBackdropTransition()\n })\n\n EventHandler.on(window, EVENT_RESIZE, () => {\n if (this._isShown && !this._isTransitioning) {\n this._adjustDialog()\n }\n })\n\n EventHandler.on(this._element, EVENT_MOUSEDOWN_DISMISS, event => {\n // a bad trick to segregate clicks that may start inside dialog but end outside, and avoid listen to scrollbar clicks\n EventHandler.one(this._element, EVENT_CLICK_DISMISS, event2 => {\n if (this._element !== event.target || this._element !== event2.target) {\n return\n }\n\n if (this._config.backdrop === 'static') {\n this._triggerBackdropTransition()\n return\n }\n\n if (this._config.backdrop) {\n this.hide()\n }\n })\n })\n }\n\n _hideModal() {\n this._element.style.display = 'none'\n this._element.setAttribute('aria-hidden', true)\n this._element.removeAttribute('aria-modal')\n this._element.removeAttribute('role')\n this._isTransitioning = false\n\n this._backdrop.hide(() => {\n document.body.classList.remove(CLASS_NAME_OPEN)\n this._resetAdjustments()\n this._scrollBar.reset()\n EventHandler.trigger(this._element, EVENT_HIDDEN)\n })\n }\n\n _isAnimated() {\n return this._element.classList.contains(CLASS_NAME_FADE)\n }\n\n _triggerBackdropTransition() {\n const hideEvent = EventHandler.trigger(this._element, EVENT_HIDE_PREVENTED)\n if (hideEvent.defaultPrevented) {\n return\n }\n\n const isModalOverflowing = this._element.scrollHeight > document.documentElement.clientHeight\n const initialOverflowY = this._element.style.overflowY\n // return if the following background transition hasn't yet completed\n if (initialOverflowY === 'hidden' || this._element.classList.contains(CLASS_NAME_STATIC)) {\n return\n }\n\n if (!isModalOverflowing) {\n this._element.style.overflowY = 'hidden'\n }\n\n this._element.classList.add(CLASS_NAME_STATIC)\n this._queueCallback(() => {\n this._element.classList.remove(CLASS_NAME_STATIC)\n this._queueCallback(() => {\n this._element.style.overflowY = initialOverflowY\n }, this._dialog)\n }, this._dialog)\n\n this._element.focus()\n }\n\n /**\n * The following methods are used to handle overflowing modals\n */\n\n _adjustDialog() {\n const isModalOverflowing = this._element.scrollHeight > document.documentElement.clientHeight\n const scrollbarWidth = this._scrollBar.getWidth()\n const isBodyOverflowing = scrollbarWidth > 0\n\n if (isBodyOverflowing && !isModalOverflowing) {\n const property = isRTL() ? 'paddingLeft' : 'paddingRight'\n this._element.style[property] = `${scrollbarWidth}px`\n }\n\n if (!isBodyOverflowing && isModalOverflowing) {\n const property = isRTL() ? 'paddingRight' : 'paddingLeft'\n this._element.style[property] = `${scrollbarWidth}px`\n }\n }\n\n _resetAdjustments() {\n this._element.style.paddingLeft = ''\n this._element.style.paddingRight = ''\n }\n\n // Static\n static jQueryInterface(config, relatedTarget) {\n return this.each(function () {\n const data = Modal.getOrCreateInstance(this, config)\n\n if (typeof config !== 'string') {\n return\n }\n\n if (typeof data[config] === 'undefined') {\n throw new TypeError(`No method named \"${config}\"`)\n }\n\n data[config](relatedTarget)\n })\n }\n}\n\n/**\n * Data API implementation\n */\n\nEventHandler.on(document, EVENT_CLICK_DATA_API, SELECTOR_DATA_TOGGLE, function (event) {\n const target = SelectorEngine.getElementFromSelector(this)\n\n if (['A', 'AREA'].includes(this.tagName)) {\n event.preventDefault()\n }\n\n EventHandler.one(target, EVENT_SHOW, showEvent => {\n if (showEvent.defaultPrevented) {\n // only register focus restorer if modal will actually get shown\n return\n }\n\n EventHandler.one(target, EVENT_HIDDEN, () => {\n if (isVisible(this)) {\n this.focus()\n }\n })\n })\n\n // avoid conflict when clicking modal toggler while another one is open\n const alreadyOpen = SelectorEngine.findOne(OPEN_SELECTOR)\n if (alreadyOpen) {\n Modal.getInstance(alreadyOpen).hide()\n }\n\n const data = Modal.getOrCreateInstance(target)\n\n data.toggle(this)\n})\n\nenableDismissTrigger(Modal)\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Modal)\n\nexport default Modal\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap offcanvas.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport SelectorEngine from './dom/selector-engine.js'\nimport Backdrop from './util/backdrop.js'\nimport { enableDismissTrigger } from './util/component-functions.js'\nimport FocusTrap from './util/focustrap.js'\nimport {\n defineJQueryPlugin,\n isDisabled,\n isVisible\n} from './util/index.js'\nimport ScrollBarHelper from './util/scrollbar.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'offcanvas'\nconst DATA_KEY = 'bs.offcanvas'\nconst EVENT_KEY = `.${DATA_KEY}`\nconst DATA_API_KEY = '.data-api'\nconst EVENT_LOAD_DATA_API = `load${EVENT_KEY}${DATA_API_KEY}`\nconst ESCAPE_KEY = 'Escape'\n\nconst CLASS_NAME_SHOW = 'show'\nconst CLASS_NAME_SHOWING = 'showing'\nconst CLASS_NAME_HIDING = 'hiding'\nconst CLASS_NAME_BACKDROP = 'offcanvas-backdrop'\nconst OPEN_SELECTOR = '.offcanvas.show'\n\nconst EVENT_SHOW = `show${EVENT_KEY}`\nconst EVENT_SHOWN = `shown${EVENT_KEY}`\nconst EVENT_HIDE = `hide${EVENT_KEY}`\nconst EVENT_HIDE_PREVENTED = `hidePrevented${EVENT_KEY}`\nconst EVENT_HIDDEN = `hidden${EVENT_KEY}`\nconst EVENT_RESIZE = `resize${EVENT_KEY}`\nconst EVENT_CLICK_DATA_API = `click${EVENT_KEY}${DATA_API_KEY}`\nconst EVENT_KEYDOWN_DISMISS = `keydown.dismiss${EVENT_KEY}`\n\nconst SELECTOR_DATA_TOGGLE = '[data-bs-toggle=\"offcanvas\"]'\n\nconst Default = {\n backdrop: true,\n keyboard: true,\n scroll: false\n}\n\nconst DefaultType = {\n backdrop: '(boolean|string)',\n keyboard: 'boolean',\n scroll: 'boolean'\n}\n\n/**\n * Class definition\n */\n\nclass Offcanvas extends BaseComponent {\n constructor(element, config) {\n super(element, config)\n\n this._isShown = false\n this._backdrop = this._initializeBackDrop()\n this._focustrap = this._initializeFocusTrap()\n this._addEventListeners()\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n toggle(relatedTarget) {\n return this._isShown ? this.hide() : this.show(relatedTarget)\n }\n\n show(relatedTarget) {\n if (this._isShown) {\n return\n }\n\n const showEvent = EventHandler.trigger(this._element, EVENT_SHOW, { relatedTarget })\n\n if (showEvent.defaultPrevented) {\n return\n }\n\n this._isShown = true\n this._backdrop.show()\n\n if (!this._config.scroll) {\n new ScrollBarHelper().hide()\n }\n\n this._element.setAttribute('aria-modal', true)\n this._element.setAttribute('role', 'dialog')\n this._element.classList.add(CLASS_NAME_SHOWING)\n\n const completeCallBack = () => {\n if (!this._config.scroll || this._config.backdrop) {\n this._focustrap.activate()\n }\n\n this._element.classList.add(CLASS_NAME_SHOW)\n this._element.classList.remove(CLASS_NAME_SHOWING)\n EventHandler.trigger(this._element, EVENT_SHOWN, { relatedTarget })\n }\n\n this._queueCallback(completeCallBack, this._element, true)\n }\n\n hide() {\n if (!this._isShown) {\n return\n }\n\n const hideEvent = EventHandler.trigger(this._element, EVENT_HIDE)\n\n if (hideEvent.defaultPrevented) {\n return\n }\n\n this._focustrap.deactivate()\n this._element.blur()\n this._isShown = false\n this._element.classList.add(CLASS_NAME_HIDING)\n this._backdrop.hide()\n\n const completeCallback = () => {\n this._element.classList.remove(CLASS_NAME_SHOW, CLASS_NAME_HIDING)\n this._element.removeAttribute('aria-modal')\n this._element.removeAttribute('role')\n\n if (!this._config.scroll) {\n new ScrollBarHelper().reset()\n }\n\n EventHandler.trigger(this._element, EVENT_HIDDEN)\n }\n\n this._queueCallback(completeCallback, this._element, true)\n }\n\n dispose() {\n this._backdrop.dispose()\n this._focustrap.deactivate()\n super.dispose()\n }\n\n // Private\n _initializeBackDrop() {\n const clickCallback = () => {\n if (this._config.backdrop === 'static') {\n EventHandler.trigger(this._element, EVENT_HIDE_PREVENTED)\n return\n }\n\n this.hide()\n }\n\n // 'static' option will be translated to true, and booleans will keep their value\n const isVisible = Boolean(this._config.backdrop)\n\n return new Backdrop({\n className: CLASS_NAME_BACKDROP,\n isVisible,\n isAnimated: true,\n rootElement: this._element.parentNode,\n clickCallback: isVisible ? clickCallback : null\n })\n }\n\n _initializeFocusTrap() {\n return new FocusTrap({\n trapElement: this._element\n })\n }\n\n _addEventListeners() {\n EventHandler.on(this._element, EVENT_KEYDOWN_DISMISS, event => {\n if (event.key !== ESCAPE_KEY) {\n return\n }\n\n if (this._config.keyboard) {\n this.hide()\n return\n }\n\n EventHandler.trigger(this._element, EVENT_HIDE_PREVENTED)\n })\n }\n\n // Static\n static jQueryInterface(config) {\n return this.each(function () {\n const data = Offcanvas.getOrCreateInstance(this, config)\n\n if (typeof config !== 'string') {\n return\n }\n\n if (data[config] === undefined || config.startsWith('_') || config === 'constructor') {\n throw new TypeError(`No method named \"${config}\"`)\n }\n\n data[config](this)\n })\n }\n}\n\n/**\n * Data API implementation\n */\n\nEventHandler.on(document, EVENT_CLICK_DATA_API, SELECTOR_DATA_TOGGLE, function (event) {\n const target = SelectorEngine.getElementFromSelector(this)\n\n if (['A', 'AREA'].includes(this.tagName)) {\n event.preventDefault()\n }\n\n if (isDisabled(this)) {\n return\n }\n\n EventHandler.one(target, EVENT_HIDDEN, () => {\n // focus on trigger when it is closed\n if (isVisible(this)) {\n this.focus()\n }\n })\n\n // avoid conflict when clicking a toggler of an offcanvas, while another is open\n const alreadyOpen = SelectorEngine.findOne(OPEN_SELECTOR)\n if (alreadyOpen && alreadyOpen !== target) {\n Offcanvas.getInstance(alreadyOpen).hide()\n }\n\n const data = Offcanvas.getOrCreateInstance(target)\n data.toggle(this)\n})\n\nEventHandler.on(window, EVENT_LOAD_DATA_API, () => {\n for (const selector of SelectorEngine.find(OPEN_SELECTOR)) {\n Offcanvas.getOrCreateInstance(selector).show()\n }\n})\n\nEventHandler.on(window, EVENT_RESIZE, () => {\n for (const element of SelectorEngine.find('[aria-modal][class*=show][class*=offcanvas-]')) {\n if (getComputedStyle(element).position !== 'fixed') {\n Offcanvas.getOrCreateInstance(element).hide()\n }\n }\n})\n\nenableDismissTrigger(Offcanvas)\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Offcanvas)\n\nexport default Offcanvas\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/sanitizer.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\n// js-docs-start allow-list\nconst ARIA_ATTRIBUTE_PATTERN = /^aria-[\\w-]*$/i\n\nexport const DefaultAllowlist = {\n // Global attributes allowed on any supplied element below.\n '*': ['class', 'dir', 'id', 'lang', 'role', ARIA_ATTRIBUTE_PATTERN],\n a: ['target', 'href', 'title', 'rel'],\n area: [],\n b: [],\n br: [],\n col: [],\n code: [],\n div: [],\n em: [],\n hr: [],\n h1: [],\n h2: [],\n h3: [],\n h4: [],\n h5: [],\n h6: [],\n i: [],\n img: ['src', 'srcset', 'alt', 'title', 'width', 'height'],\n li: [],\n ol: [],\n p: [],\n pre: [],\n s: [],\n small: [],\n span: [],\n sub: [],\n sup: [],\n strong: [],\n u: [],\n ul: []\n}\n// js-docs-end allow-list\n\nconst uriAttributes = new Set([\n 'background',\n 'cite',\n 'href',\n 'itemtype',\n 'longdesc',\n 'poster',\n 'src',\n 'xlink:href'\n])\n\n/**\n * A pattern that recognizes URLs that are safe wrt. XSS in URL navigation\n * contexts.\n *\n * Shout-out to Angular https://github.com/angular/angular/blob/15.2.8/packages/core/src/sanitization/url_sanitizer.ts#L38\n */\n// eslint-disable-next-line unicorn/better-regex\nconst SAFE_URL_PATTERN = /^(?!javascript:)(?:[a-z0-9+.-]+:|[^&:/?#]*(?:[/?#]|$))/i\n\nconst allowedAttribute = (attribute, allowedAttributeList) => {\n const attributeName = attribute.nodeName.toLowerCase()\n\n if (allowedAttributeList.includes(attributeName)) {\n if (uriAttributes.has(attributeName)) {\n return Boolean(SAFE_URL_PATTERN.test(attribute.nodeValue))\n }\n\n return true\n }\n\n // Check if a regular expression validates the attribute.\n return allowedAttributeList.filter(attributeRegex => attributeRegex instanceof RegExp)\n .some(regex => regex.test(attributeName))\n}\n\nexport function sanitizeHtml(unsafeHtml, allowList, sanitizeFunction) {\n if (!unsafeHtml.length) {\n return unsafeHtml\n }\n\n if (sanitizeFunction && typeof sanitizeFunction === 'function') {\n return sanitizeFunction(unsafeHtml)\n }\n\n const domParser = new window.DOMParser()\n const createdDocument = domParser.parseFromString(unsafeHtml, 'text/html')\n const elements = [].concat(...createdDocument.body.querySelectorAll('*'))\n\n for (const element of elements) {\n const elementName = element.nodeName.toLowerCase()\n\n if (!Object.keys(allowList).includes(elementName)) {\n element.remove()\n continue\n }\n\n const attributeList = [].concat(...element.attributes)\n const allowedAttributes = [].concat(allowList['*'] || [], allowList[elementName] || [])\n\n for (const attribute of attributeList) {\n if (!allowedAttribute(attribute, allowedAttributes)) {\n element.removeAttribute(attribute.nodeName)\n }\n }\n }\n\n return createdDocument.body.innerHTML\n}\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap util/template-factory.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport SelectorEngine from '../dom/selector-engine.js'\nimport Config from './config.js'\nimport { DefaultAllowlist, sanitizeHtml } from './sanitizer.js'\nimport { execute, getElement, isElement } from './index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'TemplateFactory'\n\nconst Default = {\n allowList: DefaultAllowlist,\n content: {}, // { selector : text , selector2 : text2 , }\n extraClass: '',\n html: false,\n sanitize: true,\n sanitizeFn: null,\n template: ''\n}\n\nconst DefaultType = {\n allowList: 'object',\n content: 'object',\n extraClass: '(string|function)',\n html: 'boolean',\n sanitize: 'boolean',\n sanitizeFn: '(null|function)',\n template: 'string'\n}\n\nconst DefaultContentType = {\n entry: '(string|element|function|null)',\n selector: '(string|element)'\n}\n\n/**\n * Class definition\n */\n\nclass TemplateFactory extends Config {\n constructor(config) {\n super()\n this._config = this._getConfig(config)\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n getContent() {\n return Object.values(this._config.content)\n .map(config => this._resolvePossibleFunction(config))\n .filter(Boolean)\n }\n\n hasContent() {\n return this.getContent().length > 0\n }\n\n changeContent(content) {\n this._checkContent(content)\n this._config.content = { ...this._config.content, ...content }\n return this\n }\n\n toHtml() {\n const templateWrapper = document.createElement('div')\n templateWrapper.innerHTML = this._maybeSanitize(this._config.template)\n\n for (const [selector, text] of Object.entries(this._config.content)) {\n this._setContent(templateWrapper, text, selector)\n }\n\n const template = templateWrapper.children[0]\n const extraClass = this._resolvePossibleFunction(this._config.extraClass)\n\n if (extraClass) {\n template.classList.add(...extraClass.split(' '))\n }\n\n return template\n }\n\n // Private\n _typeCheckConfig(config) {\n super._typeCheckConfig(config)\n this._checkContent(config.content)\n }\n\n _checkContent(arg) {\n for (const [selector, content] of Object.entries(arg)) {\n super._typeCheckConfig({ selector, entry: content }, DefaultContentType)\n }\n }\n\n _setContent(template, content, selector) {\n const templateElement = SelectorEngine.findOne(selector, template)\n\n if (!templateElement) {\n return\n }\n\n content = this._resolvePossibleFunction(content)\n\n if (!content) {\n templateElement.remove()\n return\n }\n\n if (isElement(content)) {\n this._putElementInTemplate(getElement(content), templateElement)\n return\n }\n\n if (this._config.html) {\n templateElement.innerHTML = this._maybeSanitize(content)\n return\n }\n\n templateElement.textContent = content\n }\n\n _maybeSanitize(arg) {\n return this._config.sanitize ? sanitizeHtml(arg, this._config.allowList, this._config.sanitizeFn) : arg\n }\n\n _resolvePossibleFunction(arg) {\n return execute(arg, [this])\n }\n\n _putElementInTemplate(element, templateElement) {\n if (this._config.html) {\n templateElement.innerHTML = ''\n templateElement.append(element)\n return\n }\n\n templateElement.textContent = element.textContent\n }\n}\n\nexport default TemplateFactory\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap tooltip.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport * as Popper from '@popperjs/core'\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport Manipulator from './dom/manipulator.js'\nimport { defineJQueryPlugin, execute, findShadowRoot, getElement, getUID, isRTL, noop } from './util/index.js'\nimport { DefaultAllowlist } from './util/sanitizer.js'\nimport TemplateFactory from './util/template-factory.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'tooltip'\nconst DISALLOWED_ATTRIBUTES = new Set(['sanitize', 'allowList', 'sanitizeFn'])\n\nconst CLASS_NAME_FADE = 'fade'\nconst CLASS_NAME_MODAL = 'modal'\nconst CLASS_NAME_SHOW = 'show'\n\nconst SELECTOR_TOOLTIP_INNER = '.tooltip-inner'\nconst SELECTOR_MODAL = `.${CLASS_NAME_MODAL}`\n\nconst EVENT_MODAL_HIDE = 'hide.bs.modal'\n\nconst TRIGGER_HOVER = 'hover'\nconst TRIGGER_FOCUS = 'focus'\nconst TRIGGER_CLICK = 'click'\nconst TRIGGER_MANUAL = 'manual'\n\nconst EVENT_HIDE = 'hide'\nconst EVENT_HIDDEN = 'hidden'\nconst EVENT_SHOW = 'show'\nconst EVENT_SHOWN = 'shown'\nconst EVENT_INSERTED = 'inserted'\nconst EVENT_CLICK = 'click'\nconst EVENT_FOCUSIN = 'focusin'\nconst EVENT_FOCUSOUT = 'focusout'\nconst EVENT_MOUSEENTER = 'mouseenter'\nconst EVENT_MOUSELEAVE = 'mouseleave'\n\nconst AttachmentMap = {\n AUTO: 'auto',\n TOP: 'top',\n RIGHT: isRTL() ? 'left' : 'right',\n BOTTOM: 'bottom',\n LEFT: isRTL() ? 'right' : 'left'\n}\n\nconst Default = {\n allowList: DefaultAllowlist,\n animation: true,\n boundary: 'clippingParents',\n container: false,\n customClass: '',\n delay: 0,\n fallbackPlacements: ['top', 'right', 'bottom', 'left'],\n html: false,\n offset: [0, 6],\n placement: 'top',\n popperConfig: null,\n sanitize: true,\n sanitizeFn: null,\n selector: false,\n template: '
' +\n '' +\n '' +\n '
',\n title: '',\n trigger: 'hover focus'\n}\n\nconst DefaultType = {\n allowList: 'object',\n animation: 'boolean',\n boundary: '(string|element)',\n container: '(string|element|boolean)',\n customClass: '(string|function)',\n delay: '(number|object)',\n fallbackPlacements: 'array',\n html: 'boolean',\n offset: '(array|string|function)',\n placement: '(string|function)',\n popperConfig: '(null|object|function)',\n sanitize: 'boolean',\n sanitizeFn: '(null|function)',\n selector: '(string|boolean)',\n template: 'string',\n title: '(string|element|function)',\n trigger: 'string'\n}\n\n/**\n * Class definition\n */\n\nclass Tooltip extends BaseComponent {\n constructor(element, config) {\n if (typeof Popper === 'undefined') {\n throw new TypeError('Bootstrap\\'s tooltips require Popper (https://popper.js.org)')\n }\n\n super(element, config)\n\n // Private\n this._isEnabled = true\n this._timeout = 0\n this._isHovered = null\n this._activeTrigger = {}\n this._popper = null\n this._templateFactory = null\n this._newContent = null\n\n // Protected\n this.tip = null\n\n this._setListeners()\n\n if (!this._config.selector) {\n this._fixTitle()\n }\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n enable() {\n this._isEnabled = true\n }\n\n disable() {\n this._isEnabled = false\n }\n\n toggleEnabled() {\n this._isEnabled = !this._isEnabled\n }\n\n toggle() {\n if (!this._isEnabled) {\n return\n }\n\n this._activeTrigger.click = !this._activeTrigger.click\n if (this._isShown()) {\n this._leave()\n return\n }\n\n this._enter()\n }\n\n dispose() {\n clearTimeout(this._timeout)\n\n EventHandler.off(this._element.closest(SELECTOR_MODAL), EVENT_MODAL_HIDE, this._hideModalHandler)\n\n if (this._element.getAttribute('data-bs-original-title')) {\n this._element.setAttribute('title', this._element.getAttribute('data-bs-original-title'))\n }\n\n this._disposePopper()\n super.dispose()\n }\n\n show() {\n if (this._element.style.display === 'none') {\n throw new Error('Please use show on visible elements')\n }\n\n if (!(this._isWithContent() && this._isEnabled)) {\n return\n }\n\n const showEvent = EventHandler.trigger(this._element, this.constructor.eventName(EVENT_SHOW))\n const shadowRoot = findShadowRoot(this._element)\n const isInTheDom = (shadowRoot || this._element.ownerDocument.documentElement).contains(this._element)\n\n if (showEvent.defaultPrevented || !isInTheDom) {\n return\n }\n\n // TODO: v6 remove this or make it optional\n this._disposePopper()\n\n const tip = this._getTipElement()\n\n this._element.setAttribute('aria-describedby', tip.getAttribute('id'))\n\n const { container } = this._config\n\n if (!this._element.ownerDocument.documentElement.contains(this.tip)) {\n container.append(tip)\n EventHandler.trigger(this._element, this.constructor.eventName(EVENT_INSERTED))\n }\n\n this._popper = this._createPopper(tip)\n\n tip.classList.add(CLASS_NAME_SHOW)\n\n // If this is a touch-enabled device we add extra\n // empty mouseover listeners to the body's immediate children;\n // only needed because of broken event delegation on iOS\n // https://www.quirksmode.org/blog/archives/2014/02/mouse_event_bub.html\n if ('ontouchstart' in document.documentElement) {\n for (const element of [].concat(...document.body.children)) {\n EventHandler.on(element, 'mouseover', noop)\n }\n }\n\n const complete = () => {\n EventHandler.trigger(this._element, this.constructor.eventName(EVENT_SHOWN))\n\n if (this._isHovered === false) {\n this._leave()\n }\n\n this._isHovered = false\n }\n\n this._queueCallback(complete, this.tip, this._isAnimated())\n }\n\n hide() {\n if (!this._isShown()) {\n return\n }\n\n const hideEvent = EventHandler.trigger(this._element, this.constructor.eventName(EVENT_HIDE))\n if (hideEvent.defaultPrevented) {\n return\n }\n\n const tip = this._getTipElement()\n tip.classList.remove(CLASS_NAME_SHOW)\n\n // If this is a touch-enabled device we remove the extra\n // empty mouseover listeners we added for iOS support\n if ('ontouchstart' in document.documentElement) {\n for (const element of [].concat(...document.body.children)) {\n EventHandler.off(element, 'mouseover', noop)\n }\n }\n\n this._activeTrigger[TRIGGER_CLICK] = false\n this._activeTrigger[TRIGGER_FOCUS] = false\n this._activeTrigger[TRIGGER_HOVER] = false\n this._isHovered = null // it is a trick to support manual triggering\n\n const complete = () => {\n if (this._isWithActiveTrigger()) {\n return\n }\n\n if (!this._isHovered) {\n this._disposePopper()\n }\n\n this._element.removeAttribute('aria-describedby')\n EventHandler.trigger(this._element, this.constructor.eventName(EVENT_HIDDEN))\n }\n\n this._queueCallback(complete, this.tip, this._isAnimated())\n }\n\n update() {\n if (this._popper) {\n this._popper.update()\n }\n }\n\n // Protected\n _isWithContent() {\n return Boolean(this._getTitle())\n }\n\n _getTipElement() {\n if (!this.tip) {\n this.tip = this._createTipElement(this._newContent || this._getContentForTemplate())\n }\n\n return this.tip\n }\n\n _createTipElement(content) {\n const tip = this._getTemplateFactory(content).toHtml()\n\n // TODO: remove this check in v6\n if (!tip) {\n return null\n }\n\n tip.classList.remove(CLASS_NAME_FADE, CLASS_NAME_SHOW)\n // TODO: v6 the following can be achieved with CSS only\n tip.classList.add(`bs-${this.constructor.NAME}-auto`)\n\n const tipId = getUID(this.constructor.NAME).toString()\n\n tip.setAttribute('id', tipId)\n\n if (this._isAnimated()) {\n tip.classList.add(CLASS_NAME_FADE)\n }\n\n return tip\n }\n\n setContent(content) {\n this._newContent = content\n if (this._isShown()) {\n this._disposePopper()\n this.show()\n }\n }\n\n _getTemplateFactory(content) {\n if (this._templateFactory) {\n this._templateFactory.changeContent(content)\n } else {\n this._templateFactory = new TemplateFactory({\n ...this._config,\n // the `content` var has to be after `this._config`\n // to override config.content in case of popover\n content,\n extraClass: this._resolvePossibleFunction(this._config.customClass)\n })\n }\n\n return this._templateFactory\n }\n\n _getContentForTemplate() {\n return {\n [SELECTOR_TOOLTIP_INNER]: this._getTitle()\n }\n }\n\n _getTitle() {\n return this._resolvePossibleFunction(this._config.title) || this._element.getAttribute('data-bs-original-title')\n }\n\n // Private\n _initializeOnDelegatedTarget(event) {\n return this.constructor.getOrCreateInstance(event.delegateTarget, this._getDelegateConfig())\n }\n\n _isAnimated() {\n return this._config.animation || (this.tip && this.tip.classList.contains(CLASS_NAME_FADE))\n }\n\n _isShown() {\n return this.tip && this.tip.classList.contains(CLASS_NAME_SHOW)\n }\n\n _createPopper(tip) {\n const placement = execute(this._config.placement, [this, tip, this._element])\n const attachment = AttachmentMap[placement.toUpperCase()]\n return Popper.createPopper(this._element, tip, this._getPopperConfig(attachment))\n }\n\n _getOffset() {\n const { offset } = this._config\n\n if (typeof offset === 'string') {\n return offset.split(',').map(value => Number.parseInt(value, 10))\n }\n\n if (typeof offset === 'function') {\n return popperData => offset(popperData, this._element)\n }\n\n return offset\n }\n\n _resolvePossibleFunction(arg) {\n return execute(arg, [this._element])\n }\n\n _getPopperConfig(attachment) {\n const defaultBsPopperConfig = {\n placement: attachment,\n modifiers: [\n {\n name: 'flip',\n options: {\n fallbackPlacements: this._config.fallbackPlacements\n }\n },\n {\n name: 'offset',\n options: {\n offset: this._getOffset()\n }\n },\n {\n name: 'preventOverflow',\n options: {\n boundary: this._config.boundary\n }\n },\n {\n name: 'arrow',\n options: {\n element: `.${this.constructor.NAME}-arrow`\n }\n },\n {\n name: 'preSetPlacement',\n enabled: true,\n phase: 'beforeMain',\n fn: data => {\n // Pre-set Popper's placement attribute in order to read the arrow sizes properly.\n // Otherwise, Popper mixes up the width and height dimensions since the initial arrow style is for top placement\n this._getTipElement().setAttribute('data-popper-placement', data.state.placement)\n }\n }\n ]\n }\n\n return {\n ...defaultBsPopperConfig,\n ...execute(this._config.popperConfig, [defaultBsPopperConfig])\n }\n }\n\n _setListeners() {\n const triggers = this._config.trigger.split(' ')\n\n for (const trigger of triggers) {\n if (trigger === 'click') {\n EventHandler.on(this._element, this.constructor.eventName(EVENT_CLICK), this._config.selector, event => {\n const context = this._initializeOnDelegatedTarget(event)\n context.toggle()\n })\n } else if (trigger !== TRIGGER_MANUAL) {\n const eventIn = trigger === TRIGGER_HOVER ?\n this.constructor.eventName(EVENT_MOUSEENTER) :\n this.constructor.eventName(EVENT_FOCUSIN)\n const eventOut = trigger === TRIGGER_HOVER ?\n this.constructor.eventName(EVENT_MOUSELEAVE) :\n this.constructor.eventName(EVENT_FOCUSOUT)\n\n EventHandler.on(this._element, eventIn, this._config.selector, event => {\n const context = this._initializeOnDelegatedTarget(event)\n context._activeTrigger[event.type === 'focusin' ? TRIGGER_FOCUS : TRIGGER_HOVER] = true\n context._enter()\n })\n EventHandler.on(this._element, eventOut, this._config.selector, event => {\n const context = this._initializeOnDelegatedTarget(event)\n context._activeTrigger[event.type === 'focusout' ? TRIGGER_FOCUS : TRIGGER_HOVER] =\n context._element.contains(event.relatedTarget)\n\n context._leave()\n })\n }\n }\n\n this._hideModalHandler = () => {\n if (this._element) {\n this.hide()\n }\n }\n\n EventHandler.on(this._element.closest(SELECTOR_MODAL), EVENT_MODAL_HIDE, this._hideModalHandler)\n }\n\n _fixTitle() {\n const title = this._element.getAttribute('title')\n\n if (!title) {\n return\n }\n\n if (!this._element.getAttribute('aria-label') && !this._element.textContent.trim()) {\n this._element.setAttribute('aria-label', title)\n }\n\n this._element.setAttribute('data-bs-original-title', title) // DO NOT USE IT. Is only for backwards compatibility\n this._element.removeAttribute('title')\n }\n\n _enter() {\n if (this._isShown() || this._isHovered) {\n this._isHovered = true\n return\n }\n\n this._isHovered = true\n\n this._setTimeout(() => {\n if (this._isHovered) {\n this.show()\n }\n }, this._config.delay.show)\n }\n\n _leave() {\n if (this._isWithActiveTrigger()) {\n return\n }\n\n this._isHovered = false\n\n this._setTimeout(() => {\n if (!this._isHovered) {\n this.hide()\n }\n }, this._config.delay.hide)\n }\n\n _setTimeout(handler, timeout) {\n clearTimeout(this._timeout)\n this._timeout = setTimeout(handler, timeout)\n }\n\n _isWithActiveTrigger() {\n return Object.values(this._activeTrigger).includes(true)\n }\n\n _getConfig(config) {\n const dataAttributes = Manipulator.getDataAttributes(this._element)\n\n for (const dataAttribute of Object.keys(dataAttributes)) {\n if (DISALLOWED_ATTRIBUTES.has(dataAttribute)) {\n delete dataAttributes[dataAttribute]\n }\n }\n\n config = {\n ...dataAttributes,\n ...(typeof config === 'object' && config ? config : {})\n }\n config = this._mergeConfigObj(config)\n config = this._configAfterMerge(config)\n this._typeCheckConfig(config)\n return config\n }\n\n _configAfterMerge(config) {\n config.container = config.container === false ? document.body : getElement(config.container)\n\n if (typeof config.delay === 'number') {\n config.delay = {\n show: config.delay,\n hide: config.delay\n }\n }\n\n if (typeof config.title === 'number') {\n config.title = config.title.toString()\n }\n\n if (typeof config.content === 'number') {\n config.content = config.content.toString()\n }\n\n return config\n }\n\n _getDelegateConfig() {\n const config = {}\n\n for (const [key, value] of Object.entries(this._config)) {\n if (this.constructor.Default[key] !== value) {\n config[key] = value\n }\n }\n\n config.selector = false\n config.trigger = 'manual'\n\n // In the future can be replaced with:\n // const keysWithDifferentValues = Object.entries(this._config).filter(entry => this.constructor.Default[entry[0]] !== this._config[entry[0]])\n // `Object.fromEntries(keysWithDifferentValues)`\n return config\n }\n\n _disposePopper() {\n if (this._popper) {\n this._popper.destroy()\n this._popper = null\n }\n\n if (this.tip) {\n this.tip.remove()\n this.tip = null\n }\n }\n\n // Static\n static jQueryInterface(config) {\n return this.each(function () {\n const data = Tooltip.getOrCreateInstance(this, config)\n\n if (typeof config !== 'string') {\n return\n }\n\n if (typeof data[config] === 'undefined') {\n throw new TypeError(`No method named \"${config}\"`)\n }\n\n data[config]()\n })\n }\n}\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Tooltip)\n\nexport default Tooltip\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap popover.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport Tooltip from './tooltip.js'\nimport { defineJQueryPlugin } from './util/index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'popover'\n\nconst SELECTOR_TITLE = '.popover-header'\nconst SELECTOR_CONTENT = '.popover-body'\n\nconst Default = {\n ...Tooltip.Default,\n content: '',\n offset: [0, 8],\n placement: 'right',\n template: '' +\n '' +\n '' +\n '' +\n '
',\n trigger: 'click'\n}\n\nconst DefaultType = {\n ...Tooltip.DefaultType,\n content: '(null|string|element|function)'\n}\n\n/**\n * Class definition\n */\n\nclass Popover extends Tooltip {\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Overrides\n _isWithContent() {\n return this._getTitle() || this._getContent()\n }\n\n // Private\n _getContentForTemplate() {\n return {\n [SELECTOR_TITLE]: this._getTitle(),\n [SELECTOR_CONTENT]: this._getContent()\n }\n }\n\n _getContent() {\n return this._resolvePossibleFunction(this._config.content)\n }\n\n // Static\n static jQueryInterface(config) {\n return this.each(function () {\n const data = Popover.getOrCreateInstance(this, config)\n\n if (typeof config !== 'string') {\n return\n }\n\n if (typeof data[config] === 'undefined') {\n throw new TypeError(`No method named \"${config}\"`)\n }\n\n data[config]()\n })\n }\n}\n\n/**\n * jQuery\n */\n\ndefineJQueryPlugin(Popover)\n\nexport default Popover\n","/**\n * --------------------------------------------------------------------------\n * Bootstrap scrollspy.js\n * Licensed under MIT (https://github.com/twbs/bootstrap/blob/main/LICENSE)\n * --------------------------------------------------------------------------\n */\n\nimport BaseComponent from './base-component.js'\nimport EventHandler from './dom/event-handler.js'\nimport SelectorEngine from './dom/selector-engine.js'\nimport { defineJQueryPlugin, getElement, isDisabled, isVisible } from './util/index.js'\n\n/**\n * Constants\n */\n\nconst NAME = 'scrollspy'\nconst DATA_KEY = 'bs.scrollspy'\nconst EVENT_KEY = `.${DATA_KEY}`\nconst DATA_API_KEY = '.data-api'\n\nconst EVENT_ACTIVATE = `activate${EVENT_KEY}`\nconst EVENT_CLICK = `click${EVENT_KEY}`\nconst EVENT_LOAD_DATA_API = `load${EVENT_KEY}${DATA_API_KEY}`\n\nconst CLASS_NAME_DROPDOWN_ITEM = 'dropdown-item'\nconst CLASS_NAME_ACTIVE = 'active'\n\nconst SELECTOR_DATA_SPY = '[data-bs-spy=\"scroll\"]'\nconst SELECTOR_TARGET_LINKS = '[href]'\nconst SELECTOR_NAV_LIST_GROUP = '.nav, .list-group'\nconst SELECTOR_NAV_LINKS = '.nav-link'\nconst SELECTOR_NAV_ITEMS = '.nav-item'\nconst SELECTOR_LIST_ITEMS = '.list-group-item'\nconst SELECTOR_LINK_ITEMS = `${SELECTOR_NAV_LINKS}, ${SELECTOR_NAV_ITEMS} > ${SELECTOR_NAV_LINKS}, ${SELECTOR_LIST_ITEMS}`\nconst SELECTOR_DROPDOWN = '.dropdown'\nconst SELECTOR_DROPDOWN_TOGGLE = '.dropdown-toggle'\n\nconst Default = {\n offset: null, // TODO: v6 @deprecated, keep it for backwards compatibility reasons\n rootMargin: '0px 0px -25%',\n smoothScroll: false,\n target: null,\n threshold: [0.1, 0.5, 1]\n}\n\nconst DefaultType = {\n offset: '(number|null)', // TODO v6 @deprecated, keep it for backwards compatibility reasons\n rootMargin: 'string',\n smoothScroll: 'boolean',\n target: 'element',\n threshold: 'array'\n}\n\n/**\n * Class definition\n */\n\nclass ScrollSpy extends BaseComponent {\n constructor(element, config) {\n super(element, config)\n\n // this._element is the observablesContainer and config.target the menu links wrapper\n this._targetLinks = new Map()\n this._observableSections = new Map()\n this._rootElement = getComputedStyle(this._element).overflowY === 'visible' ? null : this._element\n this._activeTarget = null\n this._observer = null\n this._previousScrollData = {\n visibleEntryTop: 0,\n parentScrollTop: 0\n }\n this.refresh() // initialize\n }\n\n // Getters\n static get Default() {\n return Default\n }\n\n static get DefaultType() {\n return DefaultType\n }\n\n static get NAME() {\n return NAME\n }\n\n // Public\n refresh() {\n this._initializeTargetsAndObservables()\n this._maybeEnableSmoothScroll()\n\n if (this._observer) {\n this._observer.disconnect()\n } else {\n this._observer = this._getNewObserver()\n }\n\n for (const section of this._observableSections.values()) {\n this._observer.observe(section)\n }\n }\n\n dispose() {\n this._observer.disconnect()\n super.dispose()\n }\n\n // Private\n _configAfterMerge(config) {\n // TODO: on v6 target should be given explicitly & remove the {target: 'ss-target'} case\n config.target = getElement(config.target) || document.body\n\n // TODO: v6 Only for backwards compatibility reasons. Use rootMargin only\n config.rootMargin = config.offset ? `${config.offset}px 0px -30%` : config.rootMargin\n\n if (typeof config.threshold === 'string') {\n config.threshold = config.threshold.split(',').map(value => Number.parseFloat(value))\n }\n\n return config\n }\n\n _maybeEnableSmoothScroll() {\n if (!this._config.smoothScroll) {\n return\n }\n\n // unregister any previous listeners\n EventHandler.off(this._config.target, EVENT_CLICK)\n\n EventHandler.on(this._config.target, EVENT_CLICK, SELECTOR_TARGET_LINKS, event => {\n const observableSection = this._observableSections.get(event.target.hash)\n if (observableSection) {\n event.preventDefault()\n const root = this._rootElement || window\n const height = observableSection.offsetTop - this._element.offsetTop\n if (root.scrollTo) {\n root.scrollTo({ top: height, behavior: 'smooth' })\n return\n }\n\n // Chrome 60 doesn't support `scrollTo`\n root.scrollTop = height\n }\n })\n }\n\n _getNewObserver() {\n const options = {\n root: this._rootElement,\n threshold: this._config.threshold,\n rootMargin: this._config.rootMargin\n }\n\n return new IntersectionObserver(entries => this._observerCallback(entries), options)\n }\n\n // The logic of selection\n _observerCallback(entries) {\n const targetElement = entry => this._targetLinks.get(`#${entry.target.id}`)\n const activate = entry => {\n this._previousScrollData.visibleEntryTop = entry.target.offsetTop\n this._process(targetElement(entry))\n }\n\n const parentScrollTop = (this._rootElement || document.documentElement).scrollTop\n const userScrollsDown = parentScrollTop >= this._previousScrollData.parentScrollTop\n this._previousScrollData.parentScrollTop = parentScrollTop\n\n for (const entry of entries) {\n if (!entry.isIntersecting) {\n this._activeTarget = null\n this._clearActiveClass(targetElement(entry))\n\n continue\n }\n\n const entryIsLowerThanPrevious = entry.target.offsetTop >= this._previousScrollData.visibleEntryTop\n // if we are scrolling down, pick the bigger offsetTop\n if (userScrollsDown && entryIsLowerThanPrevious) {\n activate(entry)\n // if parent isn't scrolled, let's keep the first visible item, breaking the iteration\n if (!parentScrollTop) {\n return\n }\n\n continue\n }\n\n // if we are scrolling up, pick the smallest offsetTop\n if (!userScrollsDown && !entryIsLowerThanPrevious) {\n activate(entry)\n }\n }\n }\n\n _initializeTargetsAndObservables() {\n this._targetLinks = new Map()\n this._observableSections = new Map()\n\n const targetLinks = SelectorEngine.find(SELECTOR_TARGET_LINKS, this._config.target)\n\n for (const anchor of targetLinks) {\n // ensure that the anchor has an id and is not disabled\n if (!anchor.hash || isDisabled(anchor)) {\n continue\n }\n\n const observableSection = SelectorEngine.findOne(decodeURI(anchor.hash), this._element)\n\n // ensure that the observableSection exists & is visible\n if (isVisible(observableSection)) {\n this._targetLinks.set(decodeURI(anchor.hash), anchor)\n this._observableSections.set(anchor.hash, observableSection)\n }\n }\n }\n\n _process(target) {\n if (this._activeTarget === target) {\n return\n }\n\n this._clearActiveClass(this._config.target)\n this._activeTarget = target\n target.classList.add(CLASS_NAME_ACTIVE)\n this._activateParents(target)\n\n EventHandler.trigger(this._element, EVENT_ACTIVATE, { relatedTarget: target })\n }\n\n _activateParents(target) {\n // Activate dropdown parents\n if (target.classList.contains(CLASS_NAME_DROPDOWN_ITEM)) {\n SelectorEngine.findOne(SELECTOR_DROPDOWN_TOGGLE, target.closest(SELECTOR_DROPDOWN))\n .classList.add(CLASS_NAME_ACTIVE)\n return\n }\n\n for (const listGroup of SelectorEngine.parents(target, SELECTOR_NAV_LIST_GROUP)) {\n // Set triggered links parents as active\n // With both - and